ХИМИЯ ОРГАНИЧЕСКАЯ,
раздел химии, изучающий соединения углерода, к которым относятся, во-первых, вещества, составляющие б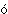 льшую часть живой материи (белки, жиры, углеводы, нуклеиновые кислоты, витамины, терпены, алкалоиды и т.д.); во-вторых, многие вещества, производимые из живых или бывших ранее живыми организмов (нефть и нефтепродукты, продукты переработки угля, пищевые продукты, шелк, хлопок, шерсть и т.д.); в-третьих, разнообразные синтетические материалы (пластмассы, краски, лекарства, синтетические волокна, красители, лабораторные и промышленные химические продукты, растворители и т.д.). Химия всех веществ, не содержащих углерода, образует второй большой раздел химии, называемый «неорганическая химия», куда входит также химия самого углерода и некоторых его простых соединений (см. также ХИМИЯ). льшую часть живой материи (белки, жиры, углеводы, нуклеиновые кислоты, витамины, терпены, алкалоиды и т.д.); во-вторых, многие вещества, производимые из живых или бывших ранее живыми организмов (нефть и нефтепродукты, продукты переработки угля, пищевые продукты, шелк, хлопок, шерсть и т.д.); в-третьих, разнообразные синтетические материалы (пластмассы, краски, лекарства, синтетические волокна, красители, лабораторные и промышленные химические продукты, растворители и т.д.). Химия всех веществ, не содержащих углерода, образует второй большой раздел химии, называемый «неорганическая химия», куда входит также химия самого углерода и некоторых его простых соединений (см. также ХИМИЯ).
Обилие органических соединений представляет собой прямой результат уникальной способности атомов углерода образовывать прочные ковалентные связи друг с другом, формируя углеродные цепи, и с такими элементами, как водород, кислород, азот, фосфор, сера и галогены (фтор, хлор, бром и иод). Каждый углеродный атом способен образовать четыре связи, которые могут быть использованы либо для формирования прямых, разветвленных или замкнутых в кольца цепей с другими углеродными атомами посредством простых, двойных или тройных связей, либо для связывания с другими элементами. Почти все органические соединения содержат также водород, и второе широко используемое определение рассматривает органическую химию как «химию углеводородов (т.е. соединений углерода с водородом) и их производных» (см. также УГЛЕРОД).
Название «органическая» ведет свое происхождение от принятой в начале 19 в. теории, согласно которой химия живых организмов, т.е. «органическая химия», каким-то уникальным образом зависит от присущей всему живому «живой силы» и эту силу якобы невозможно воспроизвести в химической лаборатории. Ошибочность этого представления была показана (1828) немецким химиком Ф.Вёлером, превратившим «неорганическое» вещество цианат аммония в «органическое» соединение мочевину:
 Начиная с этого момента органическая химия развивалась в ускоряющемся темпе. Этому способствовали структурные теории таких исследователей, как Ф.Кекуле, который в 1857 установил, что углерод в органических соединениях образует четыре связи, как и в неорганических, а в 1865 предложил структуру, объясняющую необычный характер бензола и других ароматических соединений; Я.Вант-Гофф и Ж.Ле Бель, которые предложили в 1874 тетраэдрическую модель расположения связей вокруг углерода и теорию асимметрического атома углерода; Г.Льюис, который в 1916 выдвинул концепцию ковалентной связи, состоящей из обобществленной пары электронов. Позднее для более тонкой трактовки структуры некоторые исследователи стали привлекать принципы квантовой механики. Среди них был Л.Полинг, развивший в 1930-е годы математическую концепцию резонанса (изменения электронной конфигурации в молекуле). Применяя простые структурные представления, можно подразделить сотни тысяч известных органических соединений на относительно небольшое число классов и предсказать некоторые свойства и стабильность множества еще неизученных веществ.
Начиная с этого момента органическая химия развивалась в ускоряющемся темпе. Этому способствовали структурные теории таких исследователей, как Ф.Кекуле, который в 1857 установил, что углерод в органических соединениях образует четыре связи, как и в неорганических, а в 1865 предложил структуру, объясняющую необычный характер бензола и других ароматических соединений; Я.Вант-Гофф и Ж.Ле Бель, которые предложили в 1874 тетраэдрическую модель расположения связей вокруг углерода и теорию асимметрического атома углерода; Г.Льюис, который в 1916 выдвинул концепцию ковалентной связи, состоящей из обобществленной пары электронов. Позднее для более тонкой трактовки структуры некоторые исследователи стали привлекать принципы квантовой механики. Среди них был Л.Полинг, развивший в 1930-е годы математическую концепцию резонанса (изменения электронной конфигурации в молекуле). Применяя простые структурные представления, можно подразделить сотни тысяч известных органических соединений на относительно небольшое число классов и предсказать некоторые свойства и стабильность множества еще неизученных веществ.
Для последующего обсуждения будет полезным понимание некоторых основных химических концепций, изложенных в статье ХИМИЯ.
I. КЛАССЫ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Органические соединения (углеводороды и их производные) можно разделить на два типа: ациклические (или алифатические, т.е. с открытой углеродной цепью) и циклические; последние в свою очередь подразделяются на алициклические, в молекулах которых содержатся углеродные кольца неароматического характера; ароматические, проявляющие свойства, характерные для бензола; гетероциклические, в которых один или несколько атомов в кольце представляют собой атомы неметаллов, отличные от углерода. Внутри каждого из этих типов возможна дальнейшая дифференциация на классы по присутствующим в них реакционным центрам – функциональным группам. Например, органические соединения, имеющие карбоксильную группу –СООН, являются кислотами и вступают в реакции, характерные для кислот (нейтрализация оснований, образование эфиров со спиртами и т.д.). Реакции таких групп несколько изменяются при изменении структуры молекулы, в которой они находятся; на них могут влиять и другие группы в молекуле, хотя это влияние обычно мало.
Ниже перечислены типичные функциональные группы с примерами наиболее значимых и интересных представителей каждого класса, затем рассматриваются реакции функциональных групп (разд. IV. «Реакции органических соединений»).
А. УГЛЕВОДОРОДЫ
Углеводороды являются соединениями углерода и водорода. (Простейший углеводород – метан СН4.)
Алифатические и алициклические
углеводороды могут содержать прямые (неразветвленные), разветвленные или замкнутые в кольца цепи углеродных атомов. Если четыре углеродных атома соединены один с другим только простыми (одинарными) связями, образуется углеводород бутан C4H10 с открытой (неразветвленной) цепью:
 Здесь черточки изображают ковалентные связи между углеродными атомами в плоскости страницы, пунктирные линии – связи с атомами водорода ниже, а жирные клинообразные линии – с атомами водорода выше этой плоскости. Углы между углерод-углеродными связями 109°. Эта молекула может свободно вращаться вокруг простых связей (что вообще справедливо для простых связей). Структурную формулу бутана можно написать как
Здесь черточки изображают ковалентные связи между углеродными атомами в плоскости страницы, пунктирные линии – связи с атомами водорода ниже, а жирные клинообразные линии – с атомами водорода выше этой плоскости. Углы между углерод-углеродными связями 109°. Эта молекула может свободно вращаться вокруг простых связей (что вообще справедливо для простых связей). Структурную формулу бутана можно написать как
 либо проще, CH3–CH2–CH2–CH3 или CH3CH2CH2CH3.
Для четырех углеродных атомов, связанных простыми связями, возможна также структура с разветвленной цепью:
либо проще, CH3–CH2–CH2–CH3 или CH3CH2CH2CH3.
Для четырех углеродных атомов, связанных простыми связями, возможна также структура с разветвленной цепью:
 Эта формула изображает другое соединение – изобутан, у которого иные свойства, чем у бутана (например, более низкие температуры кипения и замерзания). Заметим, что и бутан, и изобутан имеют один и тот же состав: C4H10. Такие соединения, с одинаковыми брутто-формулами, называются «изомерами». Изомерия – обычное явление в органической химии, для сложных формул возможны миллиарды изомеров.
Существуют два циклических четырехуглеродных (C4) углеводорода, содержащих только простые связи: циклобутан и метилциклопропан, оба имеют брутто-формулу C4H8:
Эта формула изображает другое соединение – изобутан, у которого иные свойства, чем у бутана (например, более низкие температуры кипения и замерзания). Заметим, что и бутан, и изобутан имеют один и тот же состав: C4H10. Такие соединения, с одинаковыми брутто-формулами, называются «изомерами». Изомерия – обычное явление в органической химии, для сложных формул возможны миллиарды изомеров.
Существуют два циклических четырехуглеродных (C4) углеводорода, содержащих только простые связи: циклобутан и метилциклопропан, оба имеют брутто-формулу C4H8:
 Насыщенные и ненасыщенные углеводороды.
Насыщенные (предельные) углеводороды – алканы (парафины) – содержат только простые (одинарные) связи между атомами углерода (например, метан, бутан, изобутан и циклобутан). Если в молекуле присутствует кратная (двойная, тройная) связь, соединение называют ненасыщенным (или непредельным) – это алкены (олефины) и алкины (ацетилены). Алканы обычно химически инертны, поскольку все валентные электроны углерода и водорода прочно связаны в сильных ковалентных связях. Метан, бутан и изобутан представляют собой алканы. Циклоалканы, представителями которых являются циклобутан и метилциклопропан, – алканы, содержащие кольцо из углеродных атомов. У ненасыщенных углеводородов реакционный центр находится по месту кратной связи. Они вступают в разнообразные химические реакции гораздо легче, чем насыщенные углеводороды.
Насыщенные и ненасыщенные углеводороды.
Насыщенные (предельные) углеводороды – алканы (парафины) – содержат только простые (одинарные) связи между атомами углерода (например, метан, бутан, изобутан и циклобутан). Если в молекуле присутствует кратная (двойная, тройная) связь, соединение называют ненасыщенным (или непредельным) – это алкены (олефины) и алкины (ацетилены). Алканы обычно химически инертны, поскольку все валентные электроны углерода и водорода прочно связаны в сильных ковалентных связях. Метан, бутан и изобутан представляют собой алканы. Циклоалканы, представителями которых являются циклобутан и метилциклопропан, – алканы, содержащие кольцо из углеродных атомов. У ненасыщенных углеводородов реакционный центр находится по месту кратной связи. Они вступают в разнообразные химические реакции гораздо легче, чем насыщенные углеводороды.
Простейший из алкенов – этилен:
 Все атомы этой молекулы лежат в одной плоскости. Вращение вокруг двойных связей невозможно, поэтому если два олефина одинакового состава различаются расположением групп относительно двойной связи, то они не идентичны и называются цис-транс-изомерами. У цис-изомеров одинаковые группы, соседние с двойной связью, расположены по одну сторону двойной связи, тогда как в транс-изомерах – по разные стороны.
Все атомы этой молекулы лежат в одной плоскости. Вращение вокруг двойных связей невозможно, поэтому если два олефина одинакового состава различаются расположением групп относительно двойной связи, то они не идентичны и называются цис-транс-изомерами. У цис-изомеров одинаковые группы, соседние с двойной связью, расположены по одну сторону двойной связи, тогда как в транс-изомерах – по разные стороны.
 Ацетилен H–CєC–H является простейшим алкином. Все его атомы лежат на прямой линии, поэтому цис-транс-изомерия невозможна, хотя атомы, связанные тройной связью, не могут вращаться друг относительно друга. Молекула бутина-2 H3C–CєC–CH3 также линейна.
Ароматические углеводороды
содержат шестичленные кольца условно с тремя двойными связями, чередующимися с тремя простыми. Простейшим соединением этого типа является бензол, имеющий структуру
Ацетилен H–CєC–H является простейшим алкином. Все его атомы лежат на прямой линии, поэтому цис-транс-изомерия невозможна, хотя атомы, связанные тройной связью, не могут вращаться друг относительно друга. Молекула бутина-2 H3C–CєC–CH3 также линейна.
Ароматические углеводороды
содержат шестичленные кольца условно с тремя двойными связями, чередующимися с тремя простыми. Простейшим соединением этого типа является бензол, имеющий структуру
 Свойства.
Углеводороды представляют собой наименее полярные из органических молекул, поскольку связи C–H почти полностью ковалентны. В результате они имеют относительно низкие температуры кипения (т. кип.) и плавления (т. пл.) по сравнению с другими органическими соединениями. Углеводороды, содержащие в цепи до четырех атомов углерода включительно, при атмосферном давлении и комнатной температуре газообразны; к ним относятся топливные газы метан CH4, этан C2H6, пропан C3H8 и бутан C4H10 (последние два обычно продают в баллонах под давлением).
Насыщенные неразветвленные углеводороды становятся твердыми при комнатной температуре, начиная с углеводорода C16. Разветвленные и ненасыщенные углеводороды являются более низкоплавкими соединениями; например, углеводород с прямой цепью гексадекан CH3(CH2)14CH3, или C16H34, плавится при 20° С, а разветвленный углеводород тетрамер 2-метилпропена (C4H8)4, или C16H32, затвердевает при –139° С, тогда как ненасыщенный углеводород гексадецин-2 CH3(CH2)12CєCCH3, или C16H30, имеет т. пл. –25° С. Циклическим соединениям свойственны более высокие температуры плавления, чем алифатическим (с открытой углеродной цепью) соединениям той же молекулярной массы; например, у 1,3-диметилциклогексана C6H10(CH3)2 т. пл. –85° С, т.е. на 19° С выше, чем у неразветвленного углеводорода октена C8H16, который имеет ту же брутто-формулу (и поэтому ту же молекулярную массу), но плавится при –104° С.
Свойства.
Углеводороды представляют собой наименее полярные из органических молекул, поскольку связи C–H почти полностью ковалентны. В результате они имеют относительно низкие температуры кипения (т. кип.) и плавления (т. пл.) по сравнению с другими органическими соединениями. Углеводороды, содержащие в цепи до четырех атомов углерода включительно, при атмосферном давлении и комнатной температуре газообразны; к ним относятся топливные газы метан CH4, этан C2H6, пропан C3H8 и бутан C4H10 (последние два обычно продают в баллонах под давлением).
Насыщенные неразветвленные углеводороды становятся твердыми при комнатной температуре, начиная с углеводорода C16. Разветвленные и ненасыщенные углеводороды являются более низкоплавкими соединениями; например, углеводород с прямой цепью гексадекан CH3(CH2)14CH3, или C16H34, плавится при 20° С, а разветвленный углеводород тетрамер 2-метилпропена (C4H8)4, или C16H32, затвердевает при –139° С, тогда как ненасыщенный углеводород гексадецин-2 CH3(CH2)12CєCCH3, или C16H30, имеет т. пл. –25° С. Циклическим соединениям свойственны более высокие температуры плавления, чем алифатическим (с открытой углеродной цепью) соединениям той же молекулярной массы; например, у 1,3-диметилциклогексана C6H10(CH3)2 т. пл. –85° С, т.е. на 19° С выше, чем у неразветвленного углеводорода октена C8H16, который имеет ту же брутто-формулу (и поэтому ту же молекулярную массу), но плавится при –104° С.
Все углеводороды горят, образуя углекислый газ и воду. Химические реакции насыщенных, ненасыщенных и ароматических соединений совершенно отличны друг от друга.
Практически важные углеводороды.
Ниже перечислены некоторые имеющие большое практическое значение углеводороды, их свойства и применения.
Метан
CH4 – бесцветный, не имеющий запаха газ с т. кип. –162° С. Он является главным компонентом природного газа, широко используемого как топливо.
Сырая нефть
– сложная смесь углеводородов (и некоторых следовых соединений) вплоть до С40. Перегонка и химическая переработка нефти дают множество промышленных углеводородов и очищенных смесей углеводородов. К продуктам, получаемым перегонкой нефти, относятся (в порядке повышения т. кип.) разбавители для красок, бензин, керосин, минеральные масла, смазочные масла и асфальт (см. НЕФТЕХИМИЧЕСКИЕ ПРОДУКТЫ;
ХИМИЯ И МЕТОДЫ ПЕРЕРАБОТКИ НЕФТИ).
Циклопропан
 – бесцветный горючий газ с т. кип. –33° С. Его используют в органическом синтезе и медицине (как анестетик).
Этилен
H2C=CH2, бесцветный газ с т. кип. –102° С. Исходный материал для синтеза ряда химических продуктов, в том числе этилового спирта и полиэтилена (см. ПЛАСТМАССЫ).
Пропилен
H3C–CH=CH2, бесцветный газ с т. кип. –48° С. Мономер полипропилена.
Бутадиен и стирол.
Бутадиен H2C=CH–CH=CH2, бесцветный горючий газ без запаха с т. кип. –4,5° С. Стирол C6H5CH=CH2, бесцветная жидкость со специфическим запахом ароматических соединений, т. кип. 146° С. Эти углеводороды служат исходными мономерами для получения ряда пластмасс и каучуков.
Бензол
C6H6, прозрачная, бесцветная, горючая жидкость с характерным запахом, т. кип. 80° С. Бензол широко используется как растворитель и исходный материал для синтеза многих органических соединений, включая взрывчатые вещества, красители и медицинские препараты.
Нафталин
C10H8 с молекулярной структурой в виде двух конденсированных колец (см. табл. 4). Это белые кристаллические пластинки с т. пл. 80° С, вещество летуче. Общеизвестно его применение в виде шариков для отпугивания моли; сырье в производстве красителей.
Ацетилен
HCєCH, бесцветный газ с т. кип. –83° С. Его применяют как горючее в ацетиленовых горелках для резки и сварки металлов, а также для синтеза многих других органических соединений.
Б. ОРГАНИЧЕСКИЕ ГАЛОГЕНИДЫ
Органические галогениды имеют общую формулу R–X, где R – стандартное обозначение для углеводородных групп, а X – атом галогена (фтора, хлора, брома или иода). Органические галогениды очень важны как исходные реагенты органического синтеза и растворители. Так же, как и углеводороды, они не очень полярны, и многие из них являются жидкостями. Некоторые из них ядовиты (например, тетрахлорид углерода), другие совершенно нетоксичны (например, фреоны, используемые в холодильниках для хранения пищевых продуктов). Хлориды гораздо дешевле бромидов и иодидов и потому находят более широкое применение в качестве растворителей.
Практически важные органические галогениды.
Тетрахлорид углерода
CCl4, бесцветная негорючая жидкость, т. кип. 77° С. Его широко используют в качестве растворителя жиров и для выведения пятен, но он довольно ядовит и вдыхание заметных количеств его паров вызывает серьезное повреждение печени. Раньше его применяли для пожаротушения, но от этого пришлось отказаться, так как при высоких температурах из него может образоваться смертельно опасный газ фосген COCl2.
Хлороформ
(трихлорметан) CHCl3, жидкость со сладковатым запахом, т. кип. 61° С. Его широко применяли как анестетик, но поскольку он ядовит, были разработаны заменяющие его анестезирующие средства. Однако его еще используют для гуманного умерщвления животных. Как и тетрахлорид углерода, хлороформ является прекрасным растворителем.
Трихлорэтилен
ClCH=CCl2, стабильная, тяжелая, ядовитая жидкость, т. кип. 87° С. Широко применяется как растворитель для химической чистки наряду с другими сходными хлорсодержащими растворителями; все они дешевы, не слишком летучи и менее токсичны, чем тетрахлорид углерода.
Дихлордифторметан
CCl2F2, бесцветный инертный газ без запаха, т. кип. –30° С. Один из низкомолекулярных хлорфторуглеводородов, называемых фреонами или хладонами, которые использовались как хладагенты и пропелленты в аэрозолях для распыления красок, инсектицидов и т.д. К фреонам относятся также хлордифторметан и хлортрифторметан. Все фреоны нетоксичны и не вызывают коррозии, но в настоящее время международные экологические соглашения предусматривают постепенную замену их другими соединениями с аналогичными физическими свойствами, поскольку фреоны, попадая в стратосферу, способствуют разрушению защитного озонового слоя Земли. См. также ХОЛОДИЛЬНАЯ ТЕХНИКА.
Тетрафторэтилен
F2C=CF2, бесцветный газ с т. кип. –78,4° С, который полимеризуется, образуя тефлон, химически инертный полимер со структурой F(CF2)nF.
Хлоропрен
H2C=CCl–CH=CH2, бесцветная жидкость, т. кип. 59,4° С. Его используют преимущественно для производства хлоропренового (неопренового) каучука. См. также КАУЧУК И РЕЗИНА.
Винилхлорид
H2C=CHCl, легко ожижаемый газ, т. кип. –13,9° С. Его применяют как хладагент и в производстве пластмасс.
Метилбромид
(бромметан) CH3Br, ядовитый газ, т. кип. 5° С. Используется для стерилизации почвы, так как в высоких концентрациях убивает все живые организмы; он безвреден для человека в низких концентрациях при кратковременном контакте.
В. СПИРТЫ
Спирты имеют общую структурную формулу ROH. Группа –OH высокополярна, и поэтому спирты гораздо более растворимы в воде и других полярных растворителях, чем углеводороды, галогениды или простые эфиры. Простые спирты, содержащие менее четырех атомов углерода, смешиваются с водой во всех соотношениях. Благодаря полярному характеру спиртов, их точки кипения также относительно высоки. Так, пропан C3H8, метилхлорид CH3Cl и этиловый спирт C2H5OH имеют приблизительно одинаковые молекулярные массы, но их т. кип. лежат соответственно при –24° С, –42° С и +78° С. Первые два соединения почти полностью нерастворимы в воде, тогда как этиловый спирт смешивается с ней в любых пропорциях. Спирты вступают в многочисленные и разнообразные реакции и поэтому имеют большое значение в качестве промежуточных соединений в синтезах. Они также являются важными промышленными растворителями.
Практически важные спирты.
Метиловый спирт
(метанол) CH3OH, бесцветная растворимая в воде жидкость, т. кип. 65° С. Когда-то его получали сухой перегонкой дерева и потому его иногда называют «древесный спирт». Теперь его производят в наибольших количествах из нефти. Метиловый спирт – это смертельный яд, и его потребление внутрь может привести к слепоте или смерти. Он представляет собой важный растворитель для полярных соединений и используется в больших количествах как антифриз. Для последней цели его летучесть является серьезным недостатком, так как требует долива по мере испарения.
Этиловый спирт
(этанол) C2H5OH, бесцветная жидкость, т. кип. 78° С, полностью растворима в воде. Именно этиловый спирт содержится в спиртных напитках и приготавливается для этой цели сбраживанием крахмала, сахара или зерна. Он также находит широкое применение как промышленный растворитель и для приготовления настоек, которые являются просто растворами в спирте. Большие количества этилового спирта получают из этилена (продукта нефтяной промышленности). Этиловый спирт служит исходным материалом для приготовления длинного списка продуктов химической промышленности: уксусной кислоты, сложных эфиров и многих других низкомолекулярных соединений. Пищевой спирт идет на изготовление алкогольных напитков, но технический этиловый спирт выпускается также в виде т.н. денатурата, непригодного для питья, но подходящего для многих других целей. Денатурирование достигается прибавлением нескольких процентов постороннего трудно удаляемого вещества. Поступающий в продажу этиловый спирт содержит около пяти процентов воды, которую нельзя удалить простой перегонкой водно-спиртовой смеси. От последних следов воды можно избавиться перегонкой с небольшим количеством бензола или другими способами, дающими безводный «абсолютный» спирт.
Изопропиловый спирт
(изопропанол) (CH3)2CH–OH, бесцветная жидкость, т. кип. 82° С. Изопропиловый спирт используется как растворитель и как промежуточное вещество для получения сложных эфиров и других продуктов химической промышленности.
Бутиловый спирт
(бутанол) CH3CH2CH2CH2OH, бесцветная, частично растворимая в воде жидкость, т. кип. 118° С. В промышленности его получают сбраживанием крахмала (зерна) или сахаров при помощи особого штамма микроорганизмов, которые продуцируют бутиловый спирт и ацетон, а не этиловый спирт. н-Бутанол (нормальный бутиловый спирт, т.е. с неразветвленной углеродной цепью) находит широкое применение в производстве растворителей для лаков.
Сивушное масло
является смесью C5-спиртов, образующихся в качестве побочного продукта при получении других спиртов посредством брожения. Более обычный способ получения C5-спиртов, используемых в качестве растворителей и для приготовления эфиров, состоит в гидролизе C5-хлоридов, продуктов хлорирования соответствующей нефтяной фракции.
Этиленгликоль
HOCH2CH2OH, прозрачная, бесцветная, сиропообразная жидкость, т. кип. 198° С. Этиленгликоль находит широкое применение как антифриз, поскольку он полностью растворим в воде, дешев, нелетуч и сильно снижает точку замерзания водных растворов (60%-ный раствор этиленгликоля в воде замерзает при –40° С). Этиленгликоль умеренно ядовит.
Глицерин
HOCH2CH(OH)CH2OH, прозрачная, сладкая на вкус, бесцветная или желтоватая сиропообразная жидкость, полностью растворимая в воде, т. кип. 290° С. Легко перевариваемый и нетоксичный, глицерин находит применение как увлажняющий и текстурирующий агент в пищевых продуктах, в косметике (кремы для рук) и некоторых медицинских препаратах. В связанном виде глицерин присутствует во всех жирах и получается в больших количествах как побочный продукт при производстве мыла. Однако этого источника недостаточно для полного удовлетворения спроса на глицерин, и потому его синтезируют из продуктов переработки нефти. Одно из его главных промышленных применений – получение взрывчатого вещества нитроглицерина O2NOCH2CH(ONO2)CH2ONO2 при обработке азотной кислотой. Динамит является смесью нитроглицерина с наполнителем (см. также ГЛИЦЕРИН).
Ментол, относительно летучее твердое вещество, т. пл. 43° С, является главным компонентом масла перечной мяты. Он обладает приятным запахом, и в малых количествах, будучи примешан к сигаретному дыму, каплям от кашля, жевательной резинке и другим продуктам, вызывает ощущение «холода» во рту. Его структурная формула – бесцветный горючий газ с т. кип. –33° С. Его используют в органическом синтезе и медицине (как анестетик).
Этилен
H2C=CH2, бесцветный газ с т. кип. –102° С. Исходный материал для синтеза ряда химических продуктов, в том числе этилового спирта и полиэтилена (см. ПЛАСТМАССЫ).
Пропилен
H3C–CH=CH2, бесцветный газ с т. кип. –48° С. Мономер полипропилена.
Бутадиен и стирол.
Бутадиен H2C=CH–CH=CH2, бесцветный горючий газ без запаха с т. кип. –4,5° С. Стирол C6H5CH=CH2, бесцветная жидкость со специфическим запахом ароматических соединений, т. кип. 146° С. Эти углеводороды служат исходными мономерами для получения ряда пластмасс и каучуков.
Бензол
C6H6, прозрачная, бесцветная, горючая жидкость с характерным запахом, т. кип. 80° С. Бензол широко используется как растворитель и исходный материал для синтеза многих органических соединений, включая взрывчатые вещества, красители и медицинские препараты.
Нафталин
C10H8 с молекулярной структурой в виде двух конденсированных колец (см. табл. 4). Это белые кристаллические пластинки с т. пл. 80° С, вещество летуче. Общеизвестно его применение в виде шариков для отпугивания моли; сырье в производстве красителей.
Ацетилен
HCєCH, бесцветный газ с т. кип. –83° С. Его применяют как горючее в ацетиленовых горелках для резки и сварки металлов, а также для синтеза многих других органических соединений.
Б. ОРГАНИЧЕСКИЕ ГАЛОГЕНИДЫ
Органические галогениды имеют общую формулу R–X, где R – стандартное обозначение для углеводородных групп, а X – атом галогена (фтора, хлора, брома или иода). Органические галогениды очень важны как исходные реагенты органического синтеза и растворители. Так же, как и углеводороды, они не очень полярны, и многие из них являются жидкостями. Некоторые из них ядовиты (например, тетрахлорид углерода), другие совершенно нетоксичны (например, фреоны, используемые в холодильниках для хранения пищевых продуктов). Хлориды гораздо дешевле бромидов и иодидов и потому находят более широкое применение в качестве растворителей.
Практически важные органические галогениды.
Тетрахлорид углерода
CCl4, бесцветная негорючая жидкость, т. кип. 77° С. Его широко используют в качестве растворителя жиров и для выведения пятен, но он довольно ядовит и вдыхание заметных количеств его паров вызывает серьезное повреждение печени. Раньше его применяли для пожаротушения, но от этого пришлось отказаться, так как при высоких температурах из него может образоваться смертельно опасный газ фосген COCl2.
Хлороформ
(трихлорметан) CHCl3, жидкость со сладковатым запахом, т. кип. 61° С. Его широко применяли как анестетик, но поскольку он ядовит, были разработаны заменяющие его анестезирующие средства. Однако его еще используют для гуманного умерщвления животных. Как и тетрахлорид углерода, хлороформ является прекрасным растворителем.
Трихлорэтилен
ClCH=CCl2, стабильная, тяжелая, ядовитая жидкость, т. кип. 87° С. Широко применяется как растворитель для химической чистки наряду с другими сходными хлорсодержащими растворителями; все они дешевы, не слишком летучи и менее токсичны, чем тетрахлорид углерода.
Дихлордифторметан
CCl2F2, бесцветный инертный газ без запаха, т. кип. –30° С. Один из низкомолекулярных хлорфторуглеводородов, называемых фреонами или хладонами, которые использовались как хладагенты и пропелленты в аэрозолях для распыления красок, инсектицидов и т.д. К фреонам относятся также хлордифторметан и хлортрифторметан. Все фреоны нетоксичны и не вызывают коррозии, но в настоящее время международные экологические соглашения предусматривают постепенную замену их другими соединениями с аналогичными физическими свойствами, поскольку фреоны, попадая в стратосферу, способствуют разрушению защитного озонового слоя Земли. См. также ХОЛОДИЛЬНАЯ ТЕХНИКА.
Тетрафторэтилен
F2C=CF2, бесцветный газ с т. кип. –78,4° С, который полимеризуется, образуя тефлон, химически инертный полимер со структурой F(CF2)nF.
Хлоропрен
H2C=CCl–CH=CH2, бесцветная жидкость, т. кип. 59,4° С. Его используют преимущественно для производства хлоропренового (неопренового) каучука. См. также КАУЧУК И РЕЗИНА.
Винилхлорид
H2C=CHCl, легко ожижаемый газ, т. кип. –13,9° С. Его применяют как хладагент и в производстве пластмасс.
Метилбромид
(бромметан) CH3Br, ядовитый газ, т. кип. 5° С. Используется для стерилизации почвы, так как в высоких концентрациях убивает все живые организмы; он безвреден для человека в низких концентрациях при кратковременном контакте.
В. СПИРТЫ
Спирты имеют общую структурную формулу ROH. Группа –OH высокополярна, и поэтому спирты гораздо более растворимы в воде и других полярных растворителях, чем углеводороды, галогениды или простые эфиры. Простые спирты, содержащие менее четырех атомов углерода, смешиваются с водой во всех соотношениях. Благодаря полярному характеру спиртов, их точки кипения также относительно высоки. Так, пропан C3H8, метилхлорид CH3Cl и этиловый спирт C2H5OH имеют приблизительно одинаковые молекулярные массы, но их т. кип. лежат соответственно при –24° С, –42° С и +78° С. Первые два соединения почти полностью нерастворимы в воде, тогда как этиловый спирт смешивается с ней в любых пропорциях. Спирты вступают в многочисленные и разнообразные реакции и поэтому имеют большое значение в качестве промежуточных соединений в синтезах. Они также являются важными промышленными растворителями.
Практически важные спирты.
Метиловый спирт
(метанол) CH3OH, бесцветная растворимая в воде жидкость, т. кип. 65° С. Когда-то его получали сухой перегонкой дерева и потому его иногда называют «древесный спирт». Теперь его производят в наибольших количествах из нефти. Метиловый спирт – это смертельный яд, и его потребление внутрь может привести к слепоте или смерти. Он представляет собой важный растворитель для полярных соединений и используется в больших количествах как антифриз. Для последней цели его летучесть является серьезным недостатком, так как требует долива по мере испарения.
Этиловый спирт
(этанол) C2H5OH, бесцветная жидкость, т. кип. 78° С, полностью растворима в воде. Именно этиловый спирт содержится в спиртных напитках и приготавливается для этой цели сбраживанием крахмала, сахара или зерна. Он также находит широкое применение как промышленный растворитель и для приготовления настоек, которые являются просто растворами в спирте. Большие количества этилового спирта получают из этилена (продукта нефтяной промышленности). Этиловый спирт служит исходным материалом для приготовления длинного списка продуктов химической промышленности: уксусной кислоты, сложных эфиров и многих других низкомолекулярных соединений. Пищевой спирт идет на изготовление алкогольных напитков, но технический этиловый спирт выпускается также в виде т.н. денатурата, непригодного для питья, но подходящего для многих других целей. Денатурирование достигается прибавлением нескольких процентов постороннего трудно удаляемого вещества. Поступающий в продажу этиловый спирт содержит около пяти процентов воды, которую нельзя удалить простой перегонкой водно-спиртовой смеси. От последних следов воды можно избавиться перегонкой с небольшим количеством бензола или другими способами, дающими безводный «абсолютный» спирт.
Изопропиловый спирт
(изопропанол) (CH3)2CH–OH, бесцветная жидкость, т. кип. 82° С. Изопропиловый спирт используется как растворитель и как промежуточное вещество для получения сложных эфиров и других продуктов химической промышленности.
Бутиловый спирт
(бутанол) CH3CH2CH2CH2OH, бесцветная, частично растворимая в воде жидкость, т. кип. 118° С. В промышленности его получают сбраживанием крахмала (зерна) или сахаров при помощи особого штамма микроорганизмов, которые продуцируют бутиловый спирт и ацетон, а не этиловый спирт. н-Бутанол (нормальный бутиловый спирт, т.е. с неразветвленной углеродной цепью) находит широкое применение в производстве растворителей для лаков.
Сивушное масло
является смесью C5-спиртов, образующихся в качестве побочного продукта при получении других спиртов посредством брожения. Более обычный способ получения C5-спиртов, используемых в качестве растворителей и для приготовления эфиров, состоит в гидролизе C5-хлоридов, продуктов хлорирования соответствующей нефтяной фракции.
Этиленгликоль
HOCH2CH2OH, прозрачная, бесцветная, сиропообразная жидкость, т. кип. 198° С. Этиленгликоль находит широкое применение как антифриз, поскольку он полностью растворим в воде, дешев, нелетуч и сильно снижает точку замерзания водных растворов (60%-ный раствор этиленгликоля в воде замерзает при –40° С). Этиленгликоль умеренно ядовит.
Глицерин
HOCH2CH(OH)CH2OH, прозрачная, сладкая на вкус, бесцветная или желтоватая сиропообразная жидкость, полностью растворимая в воде, т. кип. 290° С. Легко перевариваемый и нетоксичный, глицерин находит применение как увлажняющий и текстурирующий агент в пищевых продуктах, в косметике (кремы для рук) и некоторых медицинских препаратах. В связанном виде глицерин присутствует во всех жирах и получается в больших количествах как побочный продукт при производстве мыла. Однако этого источника недостаточно для полного удовлетворения спроса на глицерин, и потому его синтезируют из продуктов переработки нефти. Одно из его главных промышленных применений – получение взрывчатого вещества нитроглицерина O2NOCH2CH(ONO2)CH2ONO2 при обработке азотной кислотой. Динамит является смесью нитроглицерина с наполнителем (см. также ГЛИЦЕРИН).
Ментол, относительно летучее твердое вещество, т. пл. 43° С, является главным компонентом масла перечной мяты. Он обладает приятным запахом, и в малых количествах, будучи примешан к сигаретному дыму, каплям от кашля, жевательной резинке и другим продуктам, вызывает ощущение «холода» во рту. Его структурная формула
 Гераниол
C10H17OH и цитронеллол C10H19OH являются главными компонентами розового масла, которому они придают приятный запах. Они используются для приготовления духов. Существуют два изомера гераниола, отличающихся только положением наиболее удаленной от ОН-группы двойной связи. Один из этих изомеров изображается формулой
Гераниол
C10H17OH и цитронеллол C10H19OH являются главными компонентами розового масла, которому они придают приятный запах. Они используются для приготовления духов. Существуют два изомера гераниола, отличающихся только положением наиболее удаленной от ОН-группы двойной связи. Один из этих изомеров изображается формулой
 Цитронеллол отличается только отсутствием ближайшей к ОН двойной связи.
Стерины
(стеролы) – общее название твердых кристаллических спиртов стероидного ряда (см. также СТЕРОИДЫ), которые содержат ОН-группу при С-3 и алифатическую боковую цепь при С-17 в пергидроциклопентанофенантреновом скелете. Примерами стеринов являются холестерин, ситостерин и эргостерин. Стерины найдены как в растительных, так и животных тканях, где входят в состав клеточных липидных мембран. У животных холестерин служит исходным веществом для биосинтеза стероидных гормонов.
Цитронеллол отличается только отсутствием ближайшей к ОН двойной связи.
Стерины
(стеролы) – общее название твердых кристаллических спиртов стероидного ряда (см. также СТЕРОИДЫ), которые содержат ОН-группу при С-3 и алифатическую боковую цепь при С-17 в пергидроциклопентанофенантреновом скелете. Примерами стеринов являются холестерин, ситостерин и эргостерин. Стерины найдены как в растительных, так и животных тканях, где входят в состав клеточных липидных мембран. У животных холестерин служит исходным веществом для биосинтеза стероидных гормонов.
 Г. ФЕНОЛЫ
Фенолы – это ароматические спирты общей формулы R–OH, где группа R является остатком ароматического углеводорода (т.е. бензола, нафталина или родственного им циклического соединения). Фенолы – слабые кислоты.
Г. ФЕНОЛЫ
Фенолы – это ароматические спирты общей формулы R–OH, где группа R является остатком ароматического углеводорода (т.е. бензола, нафталина или родственного им циклического соединения). Фенолы – слабые кислоты.
Практически важные фенолы.
Фенол
(карбоновая кислота), кристаллическое вещество, т. пл. 42° С. Обычно фенол используют в водном растворе, причем небольшое количество воды служит для «разжижения» большого количества фенола. Он имеет сильный характерный запах и представляет собой эффективный антисептик. Во время Первой мировой войны фенол широко использовался в виде разбавленного раствора для обеззараживания ран, но в современной практике его вытеснили более действенные и менее едкие дезинфицирующие средства.
 Среди промышленных продуктов, производимых из фенола, заслуживает упоминания пикриновая кислота – желтое красящее и взрывчатое вещество, применявшееся в больших количествах в Первой мировой войне, и различные другие красители.
Креозот,
используемый для предохранения и защиты древесины, содержит фенол и другие соединения, которые задерживают рост бактерий и отпугивают насекомых. Метилфенолы (крезолы) являются наиболее значимыми среди других фенольных соединений; их применяют в производстве феноло-формальдегидных смол, дезинфицирующих средств, фунгицидов, гербицидов, азокрасителей и т.д.
Нафтолы.
a-Нафтол, бесцветное или желтоватое твердое вещество с неприятным запахом, применяется в производстве красителей и синтетических душистых соединений, т. кип. 278° С. b-Нафтол, имеющий ту же брутто-формулу C10H7OH, белое блестящее твердое вещество, т. кип. 285° С; его используют как дезинфектант и в производстве красителей, медикаментов и синтетических душистых соединений.
Среди промышленных продуктов, производимых из фенола, заслуживает упоминания пикриновая кислота – желтое красящее и взрывчатое вещество, применявшееся в больших количествах в Первой мировой войне, и различные другие красители.
Креозот,
используемый для предохранения и защиты древесины, содержит фенол и другие соединения, которые задерживают рост бактерий и отпугивают насекомых. Метилфенолы (крезолы) являются наиболее значимыми среди других фенольных соединений; их применяют в производстве феноло-формальдегидных смол, дезинфицирующих средств, фунгицидов, гербицидов, азокрасителей и т.д.
Нафтолы.
a-Нафтол, бесцветное или желтоватое твердое вещество с неприятным запахом, применяется в производстве красителей и синтетических душистых соединений, т. кип. 278° С. b-Нафтол, имеющий ту же брутто-формулу C10H7OH, белое блестящее твердое вещество, т. кип. 285° С; его используют как дезинфектант и в производстве красителей, медикаментов и синтетических душистых соединений.
 Гидрохинон
(1,4-дигидроксибензол) C6H4(OH)2, водорастворимое твердое вещество, т. кип. 285° С. Его применяют как фотографический проявитель для восстановления активированных солей серебра в черное мелкодисперсное металлическое серебро.
Эвгенол и тимол.
Эвгенол, бесцветная или желтоватая маслянистая жидкость, т. кип. 254° С; тимол, белое кристаллическое вещество с «ароматическим» запахом, т. кип. 233° С, и некоторые другие родственные фенолы широко распространены в эфирных маслах, например, масле гвоздики (эвгенол) и тимьяна.
Гидрохинон
(1,4-дигидроксибензол) C6H4(OH)2, водорастворимое твердое вещество, т. кип. 285° С. Его применяют как фотографический проявитель для восстановления активированных солей серебра в черное мелкодисперсное металлическое серебро.
Эвгенол и тимол.
Эвгенол, бесцветная или желтоватая маслянистая жидкость, т. кип. 254° С; тимол, белое кристаллическое вещество с «ароматическим» запахом, т. кип. 233° С, и некоторые другие родственные фенолы широко распространены в эфирных маслах, например, масле гвоздики (эвгенол) и тимьяна.
 Ванилин
Ванилин ,
белое кристаллическое вещество, т. кип. 285° С; слегка растворим в воде. Он является основным душистым компонентом ванили. По этой причине его широко применяют в искусственных отдушках, причем лишь небольшой концентрации достаточно, чтобы обеспечить сильный ванильный запах. Для его синтеза имеется несколько путей.
 Заметим, что ванилин содержит наравне с фенольной простую эфирную и альдегидную группы.
Д. КАРБОНОВЫЕ КИСЛОТЫ
Карбоновые кислоты имеют общую формулу R–COOH. Они являются кислотами средней силы, будучи сильнее, чем фенолы, но слабее, чем такие минеральные кислоты, как соляная, азотная или серная; как все кислоты, они имеют характерный кислый вкус. Поскольку эти кислоты высокополярны, они кипят при температурах даже более высоких, чем спирты, а простейшие кислоты (содержащие до пяти атомов углерода в цепи) растворимы в воде. Низшие кислоты – жидкие вещества, но с увеличением длины углеродной цепи повышаются и температуры плавления. Карбоновые кислоты вступают во многие важные химические реакции, а получаемые из них разнообразные продукты находят широкое применение. Многие кислоты, содержащие С=О, –OH или несколько групп –COOH, играют существенную роль в биологическом обмене веществ (см. БИОХИМИЯ).
Заметим, что ванилин содержит наравне с фенольной простую эфирную и альдегидную группы.
Д. КАРБОНОВЫЕ КИСЛОТЫ
Карбоновые кислоты имеют общую формулу R–COOH. Они являются кислотами средней силы, будучи сильнее, чем фенолы, но слабее, чем такие минеральные кислоты, как соляная, азотная или серная; как все кислоты, они имеют характерный кислый вкус. Поскольку эти кислоты высокополярны, они кипят при температурах даже более высоких, чем спирты, а простейшие кислоты (содержащие до пяти атомов углерода в цепи) растворимы в воде. Низшие кислоты – жидкие вещества, но с увеличением длины углеродной цепи повышаются и температуры плавления. Карбоновые кислоты вступают во многие важные химические реакции, а получаемые из них разнообразные продукты находят широкое применение. Многие кислоты, содержащие С=О, –OH или несколько групп –COOH, играют существенную роль в биологическом обмене веществ (см. БИОХИМИЯ).
Когда кислоты реагируют с основаниями, образуются ионные соли общей формулы RCOOM, где M – ион металла. Соли жирных кислот (алифатических карбоновых кислот с длинной цепью) называются мылами.
Практически важные карбоновые кислоты.
Муравьиная кислота
HCOOH, простейшая из карбоновых кислот. Это водорастворимая жидкость, т. кип. 100,8° С. У многих жалящих насекомых муравьиная кислота служит раздражающим компонентом яда.
Уксусная кислота
CH3COOH, бесцветная прозрачная жидкость, т. кип. 118° С, важный промышленный продукт, получаемый путем сбраживания сахаров (или этилового спирта) или синтетически из нефтепродуктов либо ацетилена. Уксус представляет собой разбавленный раствор уксусной кислоты, получаемой брожением, причем его кислый вкус – это вкус уксусной кислоты. Она используется в производстве ряда пластмасс, волокон, синтетических покрытий и является хорошим растворителем.
Масляная кислота
CH3CH2CH2COOH, т. кип. 163° С, жидкость с отвратительным запахом, обусловливающая в значительной степени неприятный запах прогорклого масла. Она находит некоторое промышленное применение как растворитель и при приготовлении бутиратов.
Стеариновая кислота
CH3(CH2)16COOH, воскообразное вещество, т. пл. 70° С. Она широко распространена в связанной форме как компонент жиров и используется вместе с парафином в производстве свечей для улучшения их горючих свойств, в приготовлении натриевой соли для производства мыла и кальциевой соли для детских присыпок, в производстве резины, смазок и для многих других целей.
Жирные кислоты
– карбоновые кислоты алифатического ряда. Алифатические кислоты с числом углеродных атомов в молекуле больше 6 называют высшими жирными кислотами (ВЖК). ВЖК могут быть природными и синтетическими. Природные ВЖК – обычные одноосновные (содержат одну карбоксильную группу в молекуле), нормального строения (неразветвленные), могут быть насыщенными или ненасыщенными. Жирные кислоты с четным числом углеродных атомов, содержащие от двух до двадцати четырех атомов углерода, можно получить из природных жиров. Компонентами этих жиров являются многочисленные ненасыщенные кислоты, наиболее обычной среди которых является олеиновая CH3–(CH2)7–CH=CH–(CH2)7–COOH, содержащая подобно стеариновой 18 атомов углерода, но имеющая одну цис-углерод-углеродную двойную связь. Как и у других ненасыщенных кислот, ее точка плавления (16° С) значительно ниже, чем у соответствующих насыщенных кислот.
Адипиновая кислота
HOOC(CH2)4COOH, как все кислоты с двумя и более группами –COOH, представляет собой твердое вещество. Она используется в многотоннажном производстве найлона.
Молочная кислота
CH3CH(OH)COOH, легко растворимое в воде вещество, плавящееся при комнатной температуре (18° С) и имеющее большое значение в биологических системах. Это она придает кислый вкус прокисшему молоку.
Лимонная кислота
HOOC–C(OH)(CH2COOH)2, водорастворимое твердое вещество без запаха, содержится в плодах всех цитрусовых. Водный раствор кислоты очень похож по вкусу на сок лимона, и потому лимонная кислота используется в разнообразных напитках из-за ее характерного приятного кислого привкуса. Она играет существенную роль в превращениях сахаров в организме.
Винная кислота
HOOCCH(OH)CH(OH)COOH, существует в трех стереоизомерных формах, называемых D-, L- и мезо-. Она присутствует в винограде и может осаждаться в виде соли в винных бочках во время брожения. Эта соль, тартрат калия-натрия, обычно известна как винный камень.
Е. АЛЬДЕГИДЫ И КЕТОНЫ
Альдегиды и кетоны имеют общие формулы R–CH=O и R–CO–R соответственно. Карбонильная группа С=О высокореакционноспособна, и поэтому альдегиды и кетоны занимают необычайно важное место в органическом синтезе многочисленных и разнообразных соединений. Некоторые члены этого класса используются в качестве ароматических добавок и душистых веществ и частично обусловливают характерные запахи некоторых растительных экстрактов и эссенций.
Практически важные альдегиды и кетоны.
Формальдегид
CH2=O, газ, т. кип. –21° С, обычно используется в виде водного раствора под названием «формалин». Газообразный формальдегид полимеризуется в твердый параформ H(OCH2)nOH, из которого регенерируется при нагревании. Как и другие низкомолекулярные альдегиды, формальдегид имеет острый запах. Формалин используют для консервации биологических материалов, а формальдегид применяют в многотоннажных процессах производства синтетических волокон и пластмасс.
Ацетон
CH3COCH3, т. кип. 56° С, приятно пахнущая жидкость с высокой растворяющей способностью для таких материалов, как нитроцеллюлоза (используемая во взрывчатых веществах и лаках) и пластмассы. Ацетон применяется как растворитель и как разбавитель для лаков. Его также используют в синтезе более сложных органических соединений.
Акролеин
CH2=CH–CH=O, т. кип. 56° С; его пары обладают раздражающим действием на слизистую. Он образуется при пиролизе («перегреве») жиров.
Бензохинон.
Желтое легко возгоняющееся твердое вещество, бензохинон является простейшим членом класса соединений, известных как «хиноны», которые содержат подобную циклическую систему. Некоторые хиноны представляют собой важные красители, другие являются пигментами растений и насекомых.
 Камфора
(камфара) представляет собой твердый летучий кетон с характерным приятным запахом. Из камфарного дерева получают L-форму, а D,L-форму производят в промышленных масштабах путем синтеза из других природных продуктов.
Ж. ПРОСТЫЕ ЭФИРЫ
Простые эфиры имеют общую структуру R–O–Rў, в которой R и Rў представляют собой углеводородные группы. Низшие члены ряда являются жидкостями, полярность их молекул низка (как и в случае углеводородов и органических галогенидов).
Практически важные простые эфиры.
Диэтиловый эфир
C2H5OC2H5, обычно называемый просто «эфир», – прозрачная, бесцветная жидкость с характерным запахом, кипящая при 35° С. Эфир используют как анестетик для наркоза, как растворитель для различных органических веществ и для химической чистки одежды.
Этиленоксид
Камфора
(камфара) представляет собой твердый летучий кетон с характерным приятным запахом. Из камфарного дерева получают L-форму, а D,L-форму производят в промышленных масштабах путем синтеза из других природных продуктов.
Ж. ПРОСТЫЕ ЭФИРЫ
Простые эфиры имеют общую структуру R–O–Rў, в которой R и Rў представляют собой углеводородные группы. Низшие члены ряда являются жидкостями, полярность их молекул низка (как и в случае углеводородов и органических галогенидов).
Практически важные простые эфиры.
Диэтиловый эфир
C2H5OC2H5, обычно называемый просто «эфир», – прозрачная, бесцветная жидкость с характерным запахом, кипящая при 35° С. Эфир используют как анестетик для наркоза, как растворитель для различных органических веществ и для химической чистки одежды.
Этиленоксид
 , т. кип. 11° С, высокореакционноспособный газ, применяемый в ряде органических синтезов, в том числе при получении этиленгликоля (первичный компонент большинства антифризов) и в производстве важного класса растворителей, известных под общим названием «целлозольвов» (R–O–CH2CH2–OH).
З. СЛОЖНЫЕ ЭФИРЫ
Сложные эфиры R–COORў рассматриваются как производные карбоновых кислот, поскольку их можно получить отщеплением молекулы воды от кислоты и спирта:
R–COOH + Rў–OH ® R–COORў + H2O
Летучие эфиры – жидкости с приятным фруктовым ароматом, прекрасные растворители для органических веществ. Некоторые эфиры обусловливают аромат душистых растений.
Практически важные сложные эфиры.
Этилацетат
CH3COOC2H5, т. кип. 77° С, находит свое главное применение как растворитель. Он образуется в реакции между этиловым спиртом и уксусной кислотой в присутствии катализатора.
Амилацетат
CH3COOCH2CH2CH2CH2CH3, т. кип. 148° С, иногда называют «банановым маслом» (которое он напоминает по запаху). Он образуется в реакции между амиловым спиртом (часто – сивушным маслом) и уксусной кислотой в присутствии катализатора. Амилацетат широко применяется как растворитель для лаков, поскольку он испаряется медленнее, чем этилацетат.
Фруктовые эфиры.
Характер многих фруктовых запахов, таких, как запахи малины, вишни, винограда и рома, отчасти обусловлен летучими эфирами, например этиловым и изоамиловым эфирами муравьиной, уксусной, масляной и валериановой кислот. Имеющиеся в продаже эссенции, имитирующие эти запахи, содержат подобные эфиры.
Винилацетат
CH2=CHOOCCH3, образуется при взаимодействии уксусной кислоты с ацетиленом в присутствии катализатора. Это важный мономер для приготовления поливинилацетатных смол, клеев и красок. , т. кип. 11° С, высокореакционноспособный газ, применяемый в ряде органических синтезов, в том числе при получении этиленгликоля (первичный компонент большинства антифризов) и в производстве важного класса растворителей, известных под общим названием «целлозольвов» (R–O–CH2CH2–OH).
З. СЛОЖНЫЕ ЭФИРЫ
Сложные эфиры R–COORў рассматриваются как производные карбоновых кислот, поскольку их можно получить отщеплением молекулы воды от кислоты и спирта:
R–COOH + Rў–OH ® R–COORў + H2O
Летучие эфиры – жидкости с приятным фруктовым ароматом, прекрасные растворители для органических веществ. Некоторые эфиры обусловливают аромат душистых растений.
Практически важные сложные эфиры.
Этилацетат
CH3COOC2H5, т. кип. 77° С, находит свое главное применение как растворитель. Он образуется в реакции между этиловым спиртом и уксусной кислотой в присутствии катализатора.
Амилацетат
CH3COOCH2CH2CH2CH2CH3, т. кип. 148° С, иногда называют «банановым маслом» (которое он напоминает по запаху). Он образуется в реакции между амиловым спиртом (часто – сивушным маслом) и уксусной кислотой в присутствии катализатора. Амилацетат широко применяется как растворитель для лаков, поскольку он испаряется медленнее, чем этилацетат.
Фруктовые эфиры.
Характер многих фруктовых запахов, таких, как запахи малины, вишни, винограда и рома, отчасти обусловлен летучими эфирами, например этиловым и изоамиловым эфирами муравьиной, уксусной, масляной и валериановой кислот. Имеющиеся в продаже эссенции, имитирующие эти запахи, содержат подобные эфиры.
Винилацетат
CH2=CHOOCCH3, образуется при взаимодействии уксусной кислоты с ацетиленом в присутствии катализатора. Это важный мономер для приготовления поливинилацетатных смол, клеев и красок.
Среди других сложных эфиров большое практическое значение имеют жиры, которые являются триэфирами глицерина и жирных кислот, а также воски – эфиры высокомолекулярных спиртов и высших жирных кислот.
И. АМИНЫ
Аминами называются производные аммиака NH3, получаемые путем замещения в нем атома H на органическую группу R; в зависимости от числа замещенных атомов водорода они могут быть первичными (RNH2), вторичными (R2NH) и третичными (R3N). (Присоединение четвертой группы R дает ряд ионных солей R4N+X–, напоминающих по структуре соли аммония, которые в противоположность аминам являются нелетучими твердыми веществами без запаха или с очень слабым запахом.) Амины представляют собой самую важную группу органических оснований: они имеют большое биологическое значение и применяются для получения разнообразных производных, включая красители и медицинские препараты.
Практически важные амины.
Триметиламин
(CH3)3N, подобно другим низкомолекулярным аминам, является газом с аммиачным, или «рыбным» запахом. Он образуется при разложении рыбы. Его получают взаимодействием метилового спирта или диметилового эфира с аммиаком; используют для производства бактерицидов, флотореагентов, кормовых добавок.
Анилин
C6H5NH2, т. кип. 184° С, бесцветная маслянистая жидкость, которая быстро становится бурой при контакте с воздухом и светом. Является исходным материалом для получения ряда анилиновых красителей, лекарственных средств (сульфамидных препаратов, ацетанилида и др.), взрывчатых веществ, анилино-формальдегидных смол, антиоксидантов, фотоматериалов и т.д.
Адреналин,
амфетамин
(бензедрин)
и эфедрин.
Адреналин – гормон,
выделяемый надпочечниками, который вызывает сужение капиллярных сосудов
и тем способствует повышению давления крови. Секреция этого гормона увеличивается
в моменты стресса, помогая организму адекватно реагировать на опасность.
Амфетамин (бензедрин, синтетический препарат) и эфедрин имеют сходное действие.

Аминокислоты
содержат как амино-, так и кислотную карбоксильную группу. Аминокислоты играют большую роль в химии живых систем. Из них особенно важны a-аминокислоты общей структуры R–CH(NH2)–COOH, которые являются строительными блоками белков.
К. ДРУГИЕ ФУНКЦИОНАЛЬНЫЕ ГРУППЫ
Азотсодержащие группы.
Кроме аминов, азот содержат различные другие функциональные группы. Амиды имеют общую структуру RC(O)NRўRўў, где Rў и Rўў – органическая группа или H; почти все они являются твердыми веществами. Нитросоединения R–NO2 представляют собой важные промежуточные соединения в органическом синтезе и взрывчатые вещества – тринитротолуол, тринитрофенол (пикриновая кислота), нитроглицерин. В нитрилах азот связан с углеродом тройной связью R–CєN; азосоединения содержат группу –N=N–, которая придает окраску многим синтетическим красителям.
Соединения серы.
Наиболее важными типами сернистых соединений являются меркаптаны (тиолы) R–SH и сульфокислоты R–SO3H, представляющие собой сильные кислоты. Низшие меркаптаны – газы или жидкости с неприятным запахом. Метилмеркаптан CH3SH – газ, используемый для придания легко распознаваемого неприятного запаха топливному газу, который сам по себе почти лишен запаха, с целью обнаружения утечек газа. Бутилмеркаптан CH3CH2CH2CH2SH – жидкость, обнаруженная в пахучих железах скунса. Соли сульфокислот с длинной углеводородной цепью используют как моющие средства; сульфаниламидные лекарственные препараты и сахарин – также производные сульфокислот.

Другая классификация.
Чтобы подчеркнуть общий источник, функции или сложные структурные особенности, органические соединения часто классифицируют по другим признакам, чем их функциональные группы. См. также АЛКАЛОИДЫ;
БИОХИМИЯ;
ЦЕЛЛЮЛОЗА;
МОЮЩИЕ СРЕДСТВА;
КРАСИТЕЛИ И КРАШЕНИЕ;
ФЕРМЕНТЫ;
ЭФИРНЫЕ МАСЛА;
ЖИРЫ И МАСЛА;
ТОПЛИВО;
ГЛИКОЗИДЫ;
КРАСКИ И ПОКРЫТИЯ;
НЕФТЕХИМИЧЕСКИЕ ПРОДУКТЫ;
ПЛАСТМАССЫ;
БЕЛКИ;
КАУЧУК И РЕЗИНА;
КРЕМНИЙОРГАНИЧЕСКИЕ ПОЛИМЕРЫ;
МЫЛО;
ВИТАМИНЫ.
II. МОЛЕКУЛЯРНАЯ СТРУКТУРА
А. ХИМИЧЕСКИЕ СВЯЗИ УГЛЕРОДА
Химическая природа углерода, промежуточная между металлами и типичными неметаллами, позволяет ему образовывать ковалентные связи с большим числом элементов, чаще всего с водородом, кислородом, азотом, галогенами, серой и фосфором. Углерод образует связи с высокой степенью ионного характера с более электроположительными металлами, но такие вещества являются высокореакционноспособными и используются как промежуточные соединения в синтезе. Углерод-углеродные связи имеют ковалентный характер и бывают простые (одинарные), двойные, тройные и ароматические (см. МОЛЕКУЛ СТРОЕНИЕ).
Ароматические системы.
Бензол – родоначальник класса ароматических соединений – имеет уникальную стабильность и вступает в химические реакции, отличные от реакций неароматических систем. Есть и другие ароматические системы, наиболее обычные из которых имеют p-орбитали, доступные для образования p-связей, на каждом атоме кольца. Пятичленные кольцевые системы с двумя сопряженными (т.е. чередующимися с простыми) двойными связями и пятым атомом, несущим неподеленную пару электронов, являются также ароматическими по своим свойствам. Ниже представлены некоторые из таких систем:

Понятие ароматичности обобщил немецкий химик Э.Хюккель. Согласно правилу Хюккеля, плоские циклические сопряженные системы с числом p-электронов, равным 4n + 2, ароматичны и стабильны, а такие же системы с числом p-электронов 4n – антиароматичны и неустойчивы.
Стабильность циклических систем.
Валентный угол (угол между связями) в ненапряженном фрагменте С–С–С составляет 109°, и кольца, в которых сохраняется это значение, более стабильны, чем те, где углы сильно отклоняются от этого значения. Напряжение, возникающее в циклических системах в результате искажения валентных углов, носит название байеровского – по имени немецкого химика А.Байера, впервые предложившего такое объяснение устойчивости насыщенных колец. Так, в трехчленных кольцах, где валентный угол составляет всего 60°, кольца сильно напряжены и легко разрываются; некоторые из их реакций напоминают реакции двойной связи С=С. Четырехчленные кольца также напряжены (валентный угол 90°), но не столь сильно. Пятичленные кольца почти плоски и их углы равны 108°; поэтому они ненапряжены и стабильны. В таких шестичленных кольцах, как циклогексан, атомы углерода не лежат в одной плоскости; такие циклы являются складчатыми, что уменьшает напряжение кольца. Пяти- и шестичленные кольца являются наиболее обычными. Большие кольца также способны снижать угловое напряжение путем образования складок, но в некоторых из них (от семи- до двенадцатичленных) атомы водорода на противоположных сторонах кольца сближаются настолько, что их отталкивание делает соединение менее стабильным (прелоговское напряжение, по имени швейцарского химика В.Прелога, открывшего этот эффект).
Таутомерия.
Если молекулу или ион можно представить в виде нескольких структур, отличающихся друг от друга только распределением электронов, эти структуры называются резонансными, причем резонансные формы не находятся в равновесии одна с другой, просто действительная электронная структура молекулы является чем-то средним между этими крайностями. Однако есть ситуации, в которых атомы перемещаются в молекуле при обычных условиях так быстро, что между различными молекулярными формами самопроизвольно устанавливается равновесие. Такое явление называется таутомерией. Примером служит равновесие между кетоном и энолом (кето-энольная таутомерия):

Здесь два соединения различаются только расположением катиона водорода и пары электронов (в p-связи). Равновесие устанавливается быстро, но сильно сдвинуто в сторону кетоформы. Следовательно, спирты со структурой –C=C–OH обычно неустойчивы и быстро превращаются в кетоформу, если нет каких-то структурных особенностей, стабилизирующих энольную форму, например в фенолах, которые при переходе в кетоформу теряли бы свой ароматический характер:

Таутомерия обычна в молекулах, которые имеют структуру –CH=X или –C=XH, где X – это S, О или N. Так, молекула H2C=C(NH2)–CH3 быстро перегруппировывается в H3C–C(=NH)–CH3, а имиды R–C(OH)=NH перегруппировываются в амиды R–C(=O)NH2. Таутомерия обычна в таких биологически важных гетероциклических системах, как барбитуровая кислота и родственные ей соединения:

Такие вещества, находящиеся в таутомерном равновесии, часто вступают в реакции, характерные для обеих форм.
Другие быстрые равновесия.
Известны и другие быстрые равновесия между молекулами с родственными структурами. Если при одном и том же углеродном атоме находятся любые две из групп OH, SH или NH2, соединение обычно неустойчиво по сравнению с двоесвязной формой:

Есть случаи, когда это равновесие сдвинуто в сторону дигидрокси-соединения. Газообразный формальдегид имеет структуру CH2=O, но в водном растворе он присоединяет молекулу воды, обретая HO–CH2–OH в качестве преобладающей формы. Хлоральгидрат Cl3CCH(OH)2 стабилен в дигидроксильной форме в результате электроноакцепторного влияния трех атомов хлора.
Б. ИЗОМЕРИЯ
Изомерия углеродной цепи.
Молекулы, которые отличаются только разветвлением углеродной цепи, называют цепными изомерами. Пример уже был дан – это изомерная пара н-бутан и изобутан.
Изомерия функциональных групп.
Молекулы с одинаковыми брутто-формулами, но различными функциональными группами являются функциональными изомерами, например этиловый спирт C2H5OH и диметиловый эфир CH3–O–CH3.
Изомерия положения.
Позиционные изомеры имеют одинаковые брутто-формулы и функциональные группы, но положения функциональных групп в их молекулах различны. Так, 1-хлорпропан CH3CH2CH2Cl и 2-хлорпропан CH3CHClCH3 являются позиционными изомерами.
Геометрическая изомерия.
Геометрические изомеры состоят из одинаковых атомов, соединенных в одной и той же последовательности, но отличаются пространственным расположением этих атомов относительно двойных связей или колец. Цис-транс-изомерия олефинов и син-анти-изомерия оксимов относятся к этому типу.

Оптическая изомерия.
Молекулы называются оптическими изомерами, когда они состоят из одинаковых атомов, соединенных одним и тем же путем, но различаются пространственным расположением этих атомов так же, как правая рука отличается от левой. Такая изомерия возможна только тогда, когда молекула асимметрична, т.е. когда она не имеет плоскости симметрии. Простейший путь к такой ситуации – присоединение четырех разных групп к атому углерода. Тогда молекула становится асимметричной и существует в двух изомерных формах. Молекулы отличаются только порядком присоединения к центральному углеродному атому, который называется асимметрическим атомом углерода или хиральным центром, так как соединен с четырьмя разными группами. Отметим, что два оптических изомера являются зеркальным отражением друг друга; они называются «энантиомерами» или «оптическими антиподами» и имеют одинаковые физические и химические свойства, за исключением того, что вращают плоскость поляризованного света в противоположных направлениях и по-разному реагируют с соединениями, которые сами являются оптическими изомерами. Изомер, который вращает плоскость поляризованного света по часовой стрелке, называют d- (от «декстро» – правый) или (+)-изомером; изомер, который вращает свет против часовой стрелки, называют l- (от «лево» – левый) или (–)-изомером. Когда в молекуле присутствует более одного асимметрического центра, максимально возможное число оптических изомеров составляет 2n, где n – число асимметрических центров. Иногда некоторые из этих изомеров оказываются идентичными, и это сокращает число оптических изомеров. Так, мезо-изомеры – это оптические изомеры, которые оптически неактивны, поскольку имеют плоскость симметрии. Оптические изомеры, которые не являются зеркальными изображениями, называются «диастереомерами»; они отличаются по физическим и химическим свойствам так же, как отличаются по ним геометрические изомеры.
Эти различия можно проиллюстрировать на примере шестиуглеродных сахаров с прямой цепью, имеющих следующую структуру: CH2OH–*CHOH–*CHOH–*CHOH–*CHOH–CHO. Здесь четыре асимметрических атома, отмеченных звездочкой, соединены каждый с четырьмя разными группами; таким образом, возможно 24, или 16, изомеров. Эти 16 изомеров составляют 8 пар энантиомеров; любая пара, не являющаяся энантиомерами, представляет собой диастереомеры. Шесть из этих 16 сахаров представлены ниже в виде т.н. проекций Фишера.

Обозначения D- и L- для энантиомеров относятся не к направлению вращения (обозначаемого d или l), а к положению OH при самом нижнем (в проекции Фишера) асимметрическом углероде: когда OH справа, изомер обозначается как D, когда слева – L. D- и L-формы глюкозы имеют одинаковые точки плавления, растворимость и т.д. С другой стороны, глюкоза и галактоза, будучи диастереомерами, имеют различные точки плавления, растворимости и т.д.
III. НОМЕНКЛАТУРА ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Для наименования органических соединений используется несколько систем, но ни одна из них не подходит для всех соединений. Сохранились многие тривиальные названия, которые либо применялись еще в начальный период органической химии и отражают источник получения или характерные качества, либо являются более новыми несистематическими названиями, которые используются по причине удобства. Так, спирт CH3OH иногда называют «древесным спиртом», потому что его когда-то получали сухой перегонкой дерева; систематическое название для этого спирта – метанол. Алкалоид морфин назван по его наркотическому действию, но в этом случае тривиальное название является единственным обычно используемым, поскольку систематическое название сложно и громоздко. Тривиальные названия часто дают промышленным продуктам, особенно в фармацевтической промышленности, где продукты продаются под патентованными названиями, причем одно и то же соединение различные фирмы могут выпустить под различными названиями. Часто используются квазисистематические названия, которые не могут адекватно описать структуру соединения без дополнительной информации. Например, инсектицид ДДТ иногда называют дихлордифенилтрихлорэтаном, чего недостаточно, чтобы написать единственную структуру этого соединения, поскольку название ничего не говорит о положении атомов хлора. Полное название для главного активного компонента – 2,2-ди(4-хлорфенил)-1,1,1-трихлорэтан.

Система ИЮПАК.
Процесс создания международной номенклатуры был начат в 1892 (Женевская номенклатура), продолжен в 1930 (Льежская номенклатура), с 1947 дальнейшее развитие связано с деятельностью комиссии ИЮПАК (Международный союз теоретической и прикладной химии) по номенклатуре органических соединений. Публиковавшиеся в разные годы правила ИЮПАК собраны в 1979 в «голубой книге» [Nomenclature of Organic Chemistry, Section A, B, C, D, E, F and H, Oxford Pergamon Press, 1979]. Своей задачей комиссия ИЮПАК считает не создание новой, единой системы номенклатуры, а упорядочение, «кодификацию», имеющейся практики. Результатом этого является сосуществование в правилах ИЮПАК нескольких номенклатурных систем, а следовательно, и нескольких допустимых названий для одного и того же вещества. Правила ИЮПАК опираются на следующие системы: заместительную, радикало-функциональную, аддитивную (соединительную), заменительную номенклатуру и т.д. В заместительной номенклатуре основой названия служит один углеводородный фрагмент, а другие рассматриваются как заместители водорода (например, (C6H5)3CH – трифенилметан). В радикало-функциональной номенклатуре в основе названия лежит название характеристической функциональной группы, определяющей химический класс соединения, к которому присоединяют наименование органического радикала (например, метиловый спирт, метилэтилкетон, метилхлорид, бутилцианид, диэтиловый эфир). В соединительной номенклатуре название составляют из нескольких равноправных частей (например, C6H5–C6H5 бифенил) или добавляя обозначения присоединенных атомов к названию основной структуры (например, 1,2,3,4-тетрагидронафталин, гидрокоричная кислота, этиленоксид, стиролдихлорид). Заменительную номенклатуру применяют при наличии неуглеродных атомов (гетероатомов) в молекулярной цепи: корни латинских названий этих атомов с окончанием «а» (а-номенклатура) присоединяют к названиям всей структуры, которая получилась бы, если бы вместо гетероатомов был углерод (например, CH3–O–CH2–CH2–NH–CH2–CH2–S–CH3 2-окса-8-тиа-5-азанонан).
Система ИЮПАК является общепризнанной в мире, и лишь адаптируется соответственно грамматике языка страны (например, однотипные заместители перечисляются в алфавитном порядке). Полный набор правил применения системы ИЮПАК ко многим менее обычным типам молекул длинен и сложен. Здесь представлено лишь основное содержание системы, но это позволяет осуществлять наименование соединений, для которых применяется система.
Названия по системе ИЮПАК образуются путем видоизменения названий насыщенных углеводородов (табл. 1). Отметим, что все эти названия оканчиваются на «ан»; это окончание характерно для всех насыщенных углеводородов – алканов. Такой ряд соединений, в котором соседние члены отличаются на одну метиленовую группу (CH2), называется гомологическим рядом.
Необходимо знать также названия замещающих
групп, которые могут находиться в разветвлениях главной углеводородной
цепи. Если из углеводорода удалить один атом водорода, полученная таким
путем группа называется углеводородным радикалом; его название получают,
заменяя в названии алкана окончание «ан» на «ил» (табл. 2).
Таблица
1. НАСЫЩЕННЫЕ УГЛЕВОДОРОДЫ С ПРЯМЫМИ ЦЕПЯМИ (НОРМАЛЬНЫЕ УГЛЕВОДОРОДЫ)
Таблица
2. НАИБОЛЕЕ ОБЫЧНЫЕ ОРГАНИЧЕСКИЕ ЗАМЕСТИТЕЛИ (РАДИКАЛЫ)
Основные правила наименования соединений по системе ИЮПАК даны ниже:
1. Находят самую длинную непрерывную
цепь углеродных атомов в молекуле. Название соответствующего углеводорода
используют как основу названия соединения.
2. Атомам (иным, чем водород) и группам
вдоль этой цепи даются наименования, и эти наименования пишут перед названием
основного углеводорода.
3. Атомы углерода основной углеводородной
цепи нумеруют последовательно, начиная с конца, выбранного так, чтобы
атомы углерода, несущие заместители, получили наиболее низкие номера.
4. Положения заместителей указывают
локантами – числами перед названиями заместителей, обозначающими
порядковые номера атомов углерода, к которым они присоединены.
5. Если имеется несколько одинаковых
групп, перед их названием ставится приставка «ди», «три», «тетра», «пента»,
«гекса» и т.д., обозначающая число присутствующих групп.
6. Двойные углерод-углеродные связи
указывают суффиксом «ен» («диен», если их две, и т.п.), а тройные –
суффиксом «ин» («диин» для двух и т.д.); при использовании этих суффиксов
окончание «ан» опускают. Положение кратных связей обозначают порядковыми
номерами углеродных атомов, подобно тому, как это делается для заместителей.
7. Все название пишется одним словом.
Несколько примеров иллюстрируют эти
правила:

Наименование
таких сложных радикалов, как CH3CHCH2Cl
в последнем примере, осуществляется
по следующим правилам:
1.
Углеродный атом со «свободной» связью получает номер 1ў. Самая длинная углеродная цепь, начиная с этого места, последовательно
нумеруется и используется для основного названия (в приведенном примере
– этан).
2. С заместителями вдоль этой цепи поступают,
как описано выше при наименовании соединений.
3. Полное название сложного радикала
заключают в скобки, чтобы избежать путаницы с номерами для остальной части
молекулы.
Названия по системе ИЮПАК и обычные
названия для нескольких часто встречающихся сложных радикалов даны в табл.
2.
Циклические углеводороды называют, прибавляя
к названию углеводорода с прямой цепью приставку «цикло». Для указания
положения заместителей атомы кольца нумеруют последовательно, начиная
с главного заместителя (табл. 3).

Отметим,
что в последнем примере углеводород  просто называют бензолом (а не 1,3,5-циклогексатриеном), а соответствующий
радикал
просто называют бензолом (а не 1,3,5-циклогексатриеном), а соответствующий
радикал
 – фенилом. – фенилом.
|
Таблица 3. НАЗВАНИЯ
НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЮЩИХСЯ ФУНКЦИОНАЛЬНЫХ (ХАРАКТЕРИСТИЧЕСКИХ)
ГРУПП ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ (ГРУППЫ ПЕРЕЧИСЛЯЮТСЯ СВЕРХУ ВНИЗ
В ПОРЯДКЕ ПОНИЖАЮЩЕГОСЯ СТАРШИНСТВА)
|
| Соединения |
Группы
|
Названия групп
|
| |
|
в префиксе
|
в суффиксе
|
| Карбоновые
кислоты |
–COOH
–(C)OOHa |
карбокси-
– |
-карбоновая
кислота
-овая кислота |
| Сульфоновые
кислоты |
–SO3H |
сульфо- |
-сульфоновая
кислота
(-сульфокислота) |
| Амиды |
–CONH2
–(C)ONH2a |
карбамоил-
– |
-карбоксамид
-амид |
| Нитрилы |
–Cє N
–(C)є Na |
циано-
– |
-карбонитрил
-нитрил |
| Альдегиды |
–CH=O
–(C)H=Oa |
формил-
оксо- |
-карбальдегид
-аль |
| Кетоны |
–(C)=O |
оксо-
(кето) |
-он |
| Спирты,
фенолы |
–OH |
гидрокси- |
-ол |
| Тиолы |
–SH |
меркапто- |
-тиол |
| Амины |
–NH2 |
амино- |
-амин |
| Простые
эфирыб |
–OAlk |
алкокси- |
– |
| Галогенопроизводныеб |
F, Cl, Br, I |
фтор-,
хлор-, бром-, иод- |
– |
| Нитрозосоединенияб |
–NO |
нитрозо- |
– |
| Нитросоединенияб |
–NO2 |
нитро- |
– |
| Диазосоединенияб |
–N2 |
диазо- |
– |
| Азидыб |
–N3 |
азидо- |
– |
|
а
Атом С, взятый в
скобки, считается частью главной углеродной цепи, а не функциональной
группы (CH3COOH
– этановая, метанкарбоновая, уксусная кислота).
б
Все эти группы в
префиксе перечисляются в алфавитном порядке в названиях соединений.
|
Более
сложным циклическим соединениям обычно дают тривиальные названия и системы
нумерации. К соединениям этого типа относятся полициклические ароматические
углеводороды (в которых бензольные кольца соединены двумя общими атомами)
и гетероциклические соединения (у которых в состав колец входят гетероатомы).
Важнейшие циклические системы и их нумерация приведены в табл. 4. Отметим,
что в гетероциклах нумерация начинается с гетероатома и производится так,
чтобы другие гетероатомы получили наименьшие номера. Наименование заместителей
в этих кольцах следует основным правилам ИЮПАК, приведенным выше.

Таблица
4. ПРИМЕРЫ ЦИКЛИЧЕСКИХ УГЛЕВОДОРОДОВ
Наиболее
широко для построения названий органических соединений правила ИЮПАК рекомендуют
использовать заместительную номенклатуру. Общая схема таких названий:
1) префиксы
– боковые цепи, затем младшие функции (см.
табл. 3) в алфавитном порядке; 2) корень
– главная цепь или цикл; 3) суффиксы
– кратные связи, главная функция. Например

Геометрическую
изомерию обозначают приставками цис-
и транс-
(см. выше).
Оптическую
изомерию обозначают символами D-,
L- или мезо
- перед названием соединения, чтобы указать ряд, к которому оно принадлежит.
Другие системы используются реже. Направление вращения плоскополяризованного
света часто указывают знаком (+) для правовращающих и знаком (–) для
левовращающих изомеров.
Для кислот, кроме их систематических
наименований, в научной литературе широко используются тривиальные названия.
Некоторые важные органические кислоты перечислены ниже (табл. 5 и 6).
Таблица
5
Таблица
6
Производные
бензола
часто имеют тривиальные
названия, которые широко распространены. Так, аминобензол называется анилином,
а гидроксибензол – фенолом. Дизамещенные бензольные кольца можно называть,
номеруя атомы углерода кольца, как это обсуждалось выше, или используя
приставки орто
(о-),
мета
(м-)
и пара
(п-)
для групп, стоящих рядом, через один или два незамещенных углерода соответственно.
Так,

(Подробнее
см. ниже
«Реакции органических соединений», разд. IV-3.A)
Промежуточные
частицы.
Во многих реакциях принимают
участие промежуточные частицы, обладающие чрезвычайно высокой реакционной
способностью. В карбанионах углерод заряжен отрицательно; карбанионы –сильные
основания, они имеют тенденцию захватывать протон или атаковать положительные
атомные центры. В карбений-ионах (карбкатионах) углерод заряжен положительно;
карбкатионы стремятся атаковать центры с высокой электронной плотностью
(атомы, олефиновые связи, ароматические системы). Карбены являются незаряженными
частицами, имеющими всего лишь шесть электронов при углероде; они вступают
в химические реакции, стремясь дополнить свой секстет до октета. Свободные
радикалы также не заряжены, но имеют неподеленный и неспаренный электрон
и также весьма реакционноспособны. Эти четыре типа реакционноспособных
частиц представлены ниже простейшими их представителями, производными
метана:

IV.
РЕАКЦИИ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
В
этом разделе кратко представлены примеры реакций и соединений из различных
классов органических веществ.
Классификация
органических соединений.
В классификации, использованной
здесь, функциональные группы расположены в порядке возрастания «степени
окисления» углерода, хотя приписывать углероду степень окисления на основании
полярности углеродных связей – несколько условная процедура, поскольку
углерод остается четырехковалентным и не приобретает и не теряет электроны
полностью. Углерод-водородные или углерод-углеродные связи почти неполярны,
и степень окисления углерода в углеводородах можно приравнять нулю. Каждая
простая ковалентная связь с более электроотрицательным элементом, например
азотом, серой, кислородом или галогеном, формально увеличивает степень
окисления на единицу, тогда как каждая связь с более электроположительным
элементом понижает ее на единицу (табл. 7).
|
Таблица 7. СТЕПЕНЬ
ОКИСЛЕНИЯ УГЛЕРОДА В ОСНОВНЫХ КЛАССАХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
|
| Степень
окисления углерода |
Тип соединения
|
Простейший стабильный
представитель класса
|
| –1 |
Металлоорганические
соединения |
CH3Na, CH3MgBr |
| 0 |
Углеводороды |
CH4 |
| +1 |
Спирты |
CH3OH |
| |
Галогениды |
CH3Cl или
другие галогены (F, Br, I) |
| |
Амины |
CH3NH2, (CH3)2NH,
(CH3)3N |
| |
Соединения
четвертичного аммония |
(CH3)4N+ |
| |
Меркаптаны |
CH3SH |
| |
Тиоэфиры
(сульфиды) |
CH3SCH3 |
| |
Дисульфиды |
CH3SSCH3 |
| |
Сульфокислоты |
CH3SO3H |
| |
Нитросоединения |
CH3NO2 |
| |
Азосоединения |
CH3–N=N–CH3 |
| +2 |
Альдегиды |
H2C=O |
| |
Кетоны |
CH3COCH3 |
| |
Оксимы |
CH2=N–OH |
| |
Дигалогениды |
CH2Cl2 (и
др. галогены) |
| |
Имины |
CH2=NH |
| |
Ацетали |
CH2(OCH3)2 |
| +3 |
Кислоты |
HCOOH |
| |
Хлорангидриды
кислот |
CH3COCl |
| |
Ангидриды
кислот |
CH3CO–O–COCH3 |
| |
Сложные
эфиры |
HCOOCH3 |
| |
Ортоэфиры |
HC(OCH3)3 |
| |
Тригалогениды |
HCCl3 (и
др. галогены) |
| |
Амиды |
HCONH2, HCONHCH3,
HCON(CH3)2 |
| |
Нитрилы |
H–Cє N |
| +4 |
Производные
угольной кислоты |
(HO–CO–OH) |
| |
Мочевина
и ее производные |
H2NCONH2 |
| |
Эфиры
хлоругольной кислоты |
CH3O–COCl |
| |
Уретаны |
CH3O–CONH2 |
| |
Эфиры
угольной кислоты |
CH3O–CO–OCH3 |
| |
Ксантаты |
CH3O–CS–SNa |
Такая
классификация оправдана тем, что реакции веществ, принадлежащих к данному
«окислительному состоянию», изменяются не слишком сильно по мере увеличения
числа углеродных атомов. Характерное поведение реагирующего атома или
группы в молекуле остается обычно неизменным. Так, реакции спиртов CH3OH,
CH3CH2OH
и
CH3CH2CH2OH
можно рассматривать как практически одинаковые. Более того, можно отметить,
что различные соединения с одинаковой степенью окисления углерода могут
быть получены друг из друга простыми реакциями замещения, например: CH3OH
+ HBr ®
CH3Br
+ H2O,
тогда как переход от одной степени окисления к другой требует окисления
[O]
или восстановления [H],
и, таким образом, использования окислителя или восстановителя:

Обычно
часть молекулы, присоединенная к данной функции, не изменяется при изменении
функции в результате замещения или окисления-восстановления. Эта неизменная
часть, часто называемая радикалом, обозначается символом R
(для алифатических или алициклических систем) или Ar
(для систем, в которых функция присоединена к ароматическому ядру). Так,
написанные выше реакции можно было бы представить в общем виде следующим
образом:

Хотя
общий принцип неизменности радикалов в органических реакциях очень помог
развитию и систематизации органической химии, его, как и все обобщения,
нужно применять с осторожностью. Исключения встречаются, например, когда
R
содержит другие функции, которые также могут быть модифицированы применяемыми
реагентами, или когда процесс замещения сопровождается «молекулярной перегруппировкой»,
например, RX + Y ®
RўY + X,
как в

Способность
предсказывать такие отклонения, а также умение подавить их или использовать
для достижения цели – одно из главных качеств хорошего химика-органика.
Таблица 8 показывает взаимоотношения
типов органических соединений и порядок обсуждения их реакций в разд.
IV
|
Таблица 8. ТИПЫ
ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
|
|
Ациклические
соединения
|
| Монофункциональные |
С–1
– Металлоорганические соединения, реактивы Гриньяра |
| |
С0
– Углеводороды: парафины, олефины, ацетилены |
| |
С1+
– Алкилгалогениды, спирты, простые эфиры, амины, нитросоединения,
меркаптаны, серусодержащие соединения |
| |
С2+
– Альдегиды, кетоны, ацетали |
| |
С3+
– Кислоты и их производные: эфиры, амиды, ангидриды |
| |
С4+
– Производные угольной кислоты: эфиры, ксантаты, карбаматы, цианаты
и тиоцианаты |
| Полифункциональные |
Многоатомные
спирты: гликоли, глицерин |
| |
Гидроксиальдегиды
и гидроксикетоны: a- и b-гидроксикарбонильные соединения |
| |
Дикетоны:
a-, b- и g-дикетоны |
| |
Кислоты
с одновалентными функциями: галогено-, гидро- кси-
(окси-) и аминокислоты |
| |
Кетокислоты
(оксокислоты): a- и b-кетокислоты |
| |
Дикарбоновые
кислоты: щавелевая, малоновая, янтарная, глутаровая,
адипиновая, ненасыщенные дикарбоновые, гидрокси- и кетодикарбоновые
кислоты |
| |
Трикарбоновые
кислоты |
|
Алициклические
соединения
|
| Моноциклические |
Природные
производные циклопентана |
| |
Природные
производные циклогексана: терпены и полиоксициклогексаны |
| |
Кольца
большего размера |
| Бициклические |
Терпены |
| |
Декалин
и сесквитерпены |
| Полициклические |
|
|
Ароматические
соединения
|
| Ряд бензола |
Углеводороды
бензольного рода |
| |
Замещенные
бензола: галогениды, нитросоединения, амины, фенолы, спирты, альдегиды,
кетоны, кислоты, сульфокислоты |
| Ряд нафталина |
a- и b-производные |
| Производные
полициклических углеводородов |
Антрацены
Фенантрены
Высшие многоядерные углеводороды |
|
Гетероциклические
соединения
|
| Пятичленные
кольца |
Один
гетероатом: фуран, кумарон, тиофен, тионафтен, пиррол,
индол |
| |
Два гетероатома:
оксазол, изоксазол, тиазол, имидазол, пиразол |
| |
Три и
более гетероатомов: фуразан, триазолы, тетразолы |
| Шестичленные
кольца |
Один
гетероатом: пираны, пиридин, хинолин, изохинолин |
| |
Два гетероатома:
пиридазин, пиримидин, пиразин |
IV-1. АЦИКЛИЧЕСКИЕ
СОЕДИНЕНИЯ
А. МОНОФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ
1.
С1–:
металлоорганические соединения.
Эти соединения обычно
получают двумя методами: а) действием активного металла (Na,
Li, Mg, Zn) на органический галогенид,
например:

или
б) действием галогенида менее активного металла на металлоорганическое
соединение более активного металла, например:

Металлоорганические
соединения обычно называют, ставя на первое место название радикала и
прибавляя к нему название металла (или соли металла), к которому присоединен
радикал, например, метилнатрий CH3Na;
диэтилцинк (C2H5)2Zn;
этилртутьхлорид C2H5HgCl.
Вообще говоря, реакционная способность этих соединений возрастает с ростом
активности металла; так, реакции алкилпроизводных цинка или ртути протекают
медленнее, чем реакции алкилпроизводных магния или натрия.
Алкилпроизводные щелочных металлов (Li,
Na, K) можно приготовить взаимодействием
свободного металла с алкилгалогенидами или с диалкилртутью:

Метод
(а) (см. выше)
можно использовать для натрия и калия только в таком инертном растворителе,
как пентан. Тонко раздробленный металл должен присутствовать в большом
избытке, а реакционную смесь необходимо очень сильно перемешивать, в противном
случае реакция

разрушает
металлалкил по мере его образования.
Для
синтетической органической химии очень ценны алкилмагнийгалогениды RMgX
(реактивы Гриньяра). Обычно их готовят непосредственным действием магния
на соответствующий органический бромид (или иодид) в эфирном растворе,
как сказано выше. Из них можно приготовить:

Реакции
литийорганических соединений RLi
очень похожи на реакции реактивов Гриньяра. Действием реактивов Гриньяра
на соответствующие галогениды цинка, кадмия или ртути можно получить как
моноалкил- (R–M–Cl),
так и диалкилпроизводные (R–M–R
) этих металлов. Из этих соединений цинк- и кадмийалкилы почти так же реакционноспособны,
как и реактив Гриньяра, хотя они слабо реагируют с более инертными карбонильными
соединениями (кетонами, сложным эфирами). Ртутьалкилы инертны в большинстве
реакций, в которые вступают реактивы Гриньяра. Они легко расщепляются только
свободными галогенами и сильными неорганическими кислотами.
Действием реактивов Гриньяра на хлориды
алюминия, олова, германия и свинца можно приготовить частично или полностью
алкилированные производные. Из них тетраэтилсвинец имел большое значение
как антидетонатор (в этилированном бензине):

2.
С0:
углеводороды.
Парафиновые
углеводороды (алканы).
Эти соединения, многие
из которых встречаются в нефти, соответствуют общей формуле CnH2n
+ 2. Поскольку в них каждая
из связей углерода имеет ковалентный характер, так как замыкается либо
на углерод, либо на водород, валентная оболочка углерода полностью насыщена,
в результате чего парафиновые углеводороды химически очень инертны.
Синтетически парафины могут быть получены
восстановлением алкилгалогенидов водородом на таких катализаторах, как
палладий на карбонате кальция, действием цинка в спирте или магния (через
RMgBr)
с последующей обработкой водой:

Их
можно также приготовить прямым восстановлением спиртов водородом на медно-хромовых
катализаторах при высоких температурах и давлениях, а также действием
иодоводорода на спирты при 180°
С или гидрированием олефинов и ацетиленов на таких катализаторах, как
палладий, платина или никель. Технически важным методом получения низших
гомологов, которые представляют собой ценное топливо, является крекинг.
В этом процессе высшие гомологи, проходя через нагретую до 500–700°
С трубку, расщепляются на более простые соединения, например:

Смесь
углеводородов, пригодных в качестве топлива, можно приготовить в промышленных
масштабах по Фишеру – Тропшу. В этом процессе смесь СО и H2
(в отношении 1:2)
пропускают над кобальтовым или никелевым катализатором при 200°
С.
Из-за
их инертности к большинству химических реагентов углеводороды не представляют
большого интереса для синтетической органической химии. На солнечном свету
они реагируют с хлором, производя хлорированные углеводороды, например:

Обычно
образуются смеси различных возможных продуктов, поэтому реакция имеет
ценность главным образом для получения растворителей, где разделение компонентов
несущественно. В некоторых случаях посредством фракционной перегонки получают
чистые продукты. При высоких температурах парафины реагируют также с азотной
кислотой, производя нитропарафины:

Образующееся
вещество, 2-нитро-2-метилпропан, является ценным растворителем. Нормальные
углеводороды часто удается изомеризовать в разветвленные углеводороды
при действии безводного хлорида алюминия. Эта реакция важна для производства
моторных топлив с низкой способностью к детонации. Мерой склонности к
детонации (преждевременному воспламенению смеси горючего и воздуха в двигателях
внутреннего сгорания) служит октановое число бензина, которое определяют
сравнением со стандартными смесями гептана (октановое число 0) и 2,2,4-триметилпентана
(т.н. «изооктана»,
октановое число 100):

Олефины
(алкены).
Эти соединения, простейшим
представителем которых является этилен H2C=CH2,
соответствуют общей формуле CnH2n.
Они содержат двойную связь
 . Такая связь
способна присоединять реакционноспособные атомы и группы, причем каждый
из участвующих в ней атомов углерода в результате образует четыре простые
связи, поэтому двойная связь называется ненасыщенной. . Такая связь
способна присоединять реакционноспособные атомы и группы, причем каждый
из участвующих в ней атомов углерода в результате образует четыре простые
связи, поэтому двойная связь называется ненасыщенной.
Олефины могут быть получены:
а) каталитическим дегидрированием парафинов
над оксидом хрома или другими катализаторами

б)
дегидрацией спиртов в присутствии серной кислоты или оксида алюминия при
высоких температурах

в)
отщеплением галогеноводорода от алкилгалогенидов при помощи сильных оснований,
например этилата калия:

(когда
может образоваться более одного олефина, преобладает наиболее разветвленный).
Олефины
вступают в следующие реакции:
а) с водородом на платиновых и сходных
с ними катализаторах, давая парафины (см.
выше);
б)
с галогенами, давая вицинальные (виц)
дигалогениды, в которых атомы галогенов присоединены к двум соседним углеродным
атомам:

в)
с пероксидом водорода, перманганатом калия или тетраоксидом осмия, давая
гликоли:

г)
с серной кислотой, давая алкилсерные кислоты, которые можно прогидролизовать
до спирта:

д)
с галогеноводородами, давая алкилгалогениды:

е)
с хлорноватистой кислотой, давая хлоргидрины:

(По
правилу Марковникова, при присоединении протонных кислот или воды к несимметричным
алкенам или алкинам атом водорода присоединяется к наиболее гидрированному
атому углерода.)
В
присутствии свободных радикалов, например радикалов, образующихся при
разложении ацетилоксида H3C–C(O)–O–O–C(O)CH3,
многие органические соединения гладко присоединяются к олефиновым связям,
например,

Простейший
олефин – этилен – применяется как регулятор роста растений,
ускоряющий созревание плодов (в том числе цитрусовых), и как исходное соединение
в производстве полиэтилена, полиэтилен-пропиленовых каучуков, а также
для синтеза этиленгликоля CH2OH–CH2OH.
Во время Первой мировой войны его широко применяли для производства иприта
(горчичного газа) ClCH2CH2SCH2CH2Cl,
который получают действием дихлорида серы на этилен.
См. также
ХИМИЧЕСКОЕ И БИОЛОГИЧЕСКОЕ ОРУЖИЕ.
Многие
диены, т.е. углеводороды, содержащие две двойные связи, представляют промышленный
интерес. Бутадиен H2C=CH–CH=CH2,
хлоропрен H2C=CCl–CH=CH2
и изопрен H2C=C(CH3)CH=CH2
полимеризуются, давая каучук. Природный каучук также можно рассматривать
как полиизопреноиды природного происхождения (см.
разд. IV-1.А.4
и IV-2.Б.1).
См. также
КАУЧУК И РЕЗИНА.
Ацетилены.
Карбид кальция CaC2
при обработке водой выделяет газ ацетилен C2H2,
имеющий структуру H–CєC–H.
Это вещество является первым членом гомологического ряда ацетиленовых
углеводородов CnH2n
– 2. Наиболее общий путь получения соединений этого ряда состоит в присоединении
брома к соответствующим олефинам с последующей обработкой спиртовым раствором
гидроксида калия.
Присоединением воды в присутствии сульфата
ртути и серной кислоты ацетилен превращается в уксусный альдегид, из которого
можно получить уксусную кислоту и другие ценные технические продукты:

(Соединения
со структурой  неустойчивы, так как самопроизвольно перегруппировываются в
неустойчивы, так как самопроизвольно перегруппировываются в  .)
Ацетилен и те из его гомологов, у которых имеется водород при связанном
тройной связью углероде, ведут себя как очень слабые кислоты. Их соли
со щелочными металлами можно получить действием амида натрия или амида
калия: .)
Ацетилен и те из его гомологов, у которых имеется водород при связанном
тройной связью углероде, ведут себя как очень слабые кислоты. Их соли
со щелочными металлами можно получить действием амида натрия или амида
калия:

С
аммиачным раствором серебра или одновалентной меди ацетилены образуют
нерастворимые взрывчатые соли серебра и одновалентной меди. У всех соединений
ряда тройная связь способна присоединять реагенты подобно двойной связи.
3.
Окислительное состояние С+.
Алкилгалогениды
можно рассматривать как
производные углеводородов, у которых водород заменен галогеном. Они имеют
общую формулу R–X,
где X
может быть F, Cl, Br или
I.
Прямое
замещение водорода галогеном редко может служить препаративным методом
получения алкилгалогенидов (см.
выше). Более подходящий метод
состоит в обработке соответствующего спирта (ROH)
галогеноводородом или галогенидом фосфора, чтобы заменить гидроксильную
группу на галоген.
По многим физическим свойствам, таким,
как низкие температуры кипения и плавления, алкилгалогениды напоминают
углеводороды, поскольку оба класса соединений относительно неполярны.
Химически алкилгалогениды гораздо более реакционноспособны, причем иодиды
наиболее активны, а хлориды – наименее. По реакционной способности,
реакциям и методам получения алкилфториды сильно отличаются от других
галогенидов. Получение металлалкилов, например реактивов Гриньяра, уже
обсуждалось в разд. IV-1.А.1.
Атом галогена можно также заменить на самые разнообразные простые неорганические
или органические основания, например:

Такие
реакции замещения лучше идут с алкилбромидами и алкилиодидами, хлор в
алкилхлоридах заменить труднее.
Параллельно
с написанными выше идут побочные процессы – реакция с растворителем
и отщепление HX
с образованием олефина. Природа R
оказывает сильнейшее влияние на скорость и состав продуктов реакции.
Реакции замещения могут протекать по
двум различным механизмам: мономолекулярному (SN1)
или бимолекулярному (SN2).
Согласно первому механизму сначала происходит диссоциация алкилгалогенида
на галогенид-анион и ион карбения – нестабильную высокореакционноспособную
частицу, которая немедленно реагирует с добавленным основанием или молекулой
растворителя. Поскольку стабильность карбениевых ионов растет от первичных
к третичным

этот
механизм замещения должен быть преобладающим для третичных алкилгалогенидов
R3CX,
его роль должна снижаться для вторичных алкилгалогенидов R2CHX.
По второму механизму вступающая группа постепенно вытесняет уходящую,
причем в переходном состоянии обе группы связаны с углеродом в реакционном
центре приблизительно одинаково. Наиболее энергетически выгодным направлением
атаки для вступающей группы является подход со стороны, обратной направлению,
в котором удаляется входящая группа:

Поскольку
в переходном состоянии электронная плотность на реакционном центре выше,
чем в исходном и конечном, скорость такого процесса должна падать в ряду
RCH2X
> R2CHX
> R3CX,
т.е. в последовательности, обратной той, которая характерна для SN1-реакций.
Реальные
процессы замещения являются чем-то промежуточным по отношению к двум описанным
крайним идеальным случаям, причем реальный механизм замещения для первичных
алкилгалогенидов RCH2X
будет близок к SN2,
а для третичных алкилгалогенидов R3CX
– к SN1,
тогда как для вторичных алкилгалогенидов R2CHX
реальный механизм будет представлять собой нечто среднее. Поэтому наблюдаемые
скорости замещения обычно уменьшаются при переходе от RCH2X
к R2CHX.
Одновременно при переходе от первичных алкилгалогенидов ко вторичным и
третичным возрастает роль упомянутых выше побочных процессов – реакций
с растворителем (водой, спиртом и т.п.) и образования олефинов, которые
в случае некоторых вторичных и особенно третичных алкилгалогенидов могут
стать преобладающими.
Присутствие двойной связи вблизи галогена
также сильно изменяет реакционную способность. Так, винилгалогениды R–CH=CH–X
и арилгалогениды ArX
исключительно малоактивны; наоборот, аллилгалогениды R–CH=CH–CH2X
и бензилгалогениды Ar–CH2X
необычайно реакционноспособны.
Все перечисленные выше реагенты являются
основаниями Льюиса и могут вызывать конкурирующую реакцию отщепления,
в которой отщепляется галогеноводород и образуется олефин:

Эта
реакция идет особенно хорошо с такими сильными основаниями, как OH–
и RO–,
и становится преобладающей, когда используются третичные галогениды или
если реагентами являются спиртовые растворы сильных оснований:

Простые
полихлорированные углеводороды широко применяются в промышленности в качестве
растворителей. Среди наиболее важных растворителей можно упомянуть хлороформ
CHCl3,
дихлорэтан ClCH2CH2Cl
и тетрахлорэтан Cl2CHCHCl2.
Спирты
и простые эфиры. Одноатомные
спирты имеют общую формулу R–OH,
представляющую углеводород, в котором водород заменен гидроксильной группой.
Далее они могут быть подразделены на первичные RCH2OH,
вторичные RRўCHOH
и третичные спирты, RRўRўўCOH,
в зависимости от того, одна, две или три алкильные группы присоединены
к углероду, несущему гидроксильную группу.
Низшие спирты находят широкое применение
в промышленности в качестве растворителей и как промежуточные вещества
для синтеза. Метанол (т. кип. 64,7°
С) получают взаимодействием CO
и H2
при высоком давлении над хромо-цинковым оксидным катализатором при 350–400°
С. Этанол (обычный этиловый спирт, т. кип. 78,3°
С) традиционно получают сбраживанием сахара или крахмала в присутствии
дрожжей, хотя некоторое количество его производят путем поглощения этилена
серной кислотой с последующим гидролизом образующейся этилсерной кислоты
C2H5OSO3H
водой. Оба процесса дают разбавленные спиртовые растворы, из которых получают
перегонкой поступающий в продажу 95%-й спирт. Изопропиловый спирт (пропанол-2,
т. кип. 82,3°
С) обычно делают сернокислотным методом из пропилена CH3CH=CH2,
побочного продукта производства бензина крекингом. Он находил некоторое
применение как заменитель этанола в качестве растворителя и в спиртовых
растираниях. Некоторые из высших спиртов, например 2-этилгексанол-1 (или
«612»), действуют на насекомых как репелленты.
Общие методы лабораторного получения
спиртов включают а) гидролиз алкилгалогенидов; б) гидратацию олефинов
в присутствии минеральных кислот, например описанным выше сернокислотным
методом; в) действие реактивов Гриньяра RMgX
на альдегиды Rў–CHO
и кетоны Rў–CO–Rўў.
Формальдегид дает первичные спирты, альдегиды – вторичные спирты,
а кетоны – третичные:

Спирты
обнаруживают свойства очень слабых кислот. Водород гидроксильной группы
в спиртах несколько менее кислый, чем водород воды. Он может быть замещен
на активные металлы с образованием алкоголятов:

Эта
реакция легче всего протекает с первичными спиртами и медленнее –
с третичными. Na
реагирует очень медленно с трет-бутиловым
спиртом, но K
(более активный) реагирует быстро. Вообще реакции спиртов, в которых рвется
O–H-связь,
легче всего протекают с первичными спиртами и медленнее всего –
с третичными.
Сложные
эфиры можно получить следующим образом:

Образование
сложных эфиров по первым двум из этих реакций идет быстро и необратимо
и, как правило, не требует катализаторов (хотя обычно к реакционной смеси
прибавляют такие основания, как пиридин или триэтиламин, которые связывают
образующиеся кислоты в виде солей). Третий метод основан на обратимой
равновесной реакции и требует катализатора, обычно кислоты (этерификация
по Фишеру). Так, реакцию можно заставить протекать слева направо (гидролиз),
если использовать избыток спирта и удалять воду по мере ее образования.
Ни один из трех указанных выше методов не применим к третичным спиртам.
Спирты,
однако, амфотерны и в присутствии сильных кислот ведут себя как очень
слабые основания:

Способность
к замещению группы –OH
в этой и других реакциях убывает от третичных к первичным спиртам. В присутствии
таких дегидратирующих агентов, как серная или фосфорная кислота, при более
низких температурах из спиртов образуются простые эфиры R–O–R,
тогда как при более высоких температурах путем отщепления воды получаются
олефины. Этот метод не годится для получения простых эфиров из вторичных
спиртов, а с третичными дает только олефины. Дегидратация спиртов с образованием
олефинов может быть осуществлена каталитически в паровой фазе над такими
оксидами металлов, как оксид алюминия.
Окисление
спиртов можно осуществить при помощи сильных окислителей (хромовая или
азотная кислота). Продукты окисления различны по своей природе для первичных,
вторичных и третичных спиртов. Так, первичные спирты сначала окисляются
в альдегиды, которые, если их немедленно не удалить из окислительной среды,
окисляются далее до кислот:

Вторичные
спирты окисляются до устойчивых кетонов RCORў,
тогда как третичные спирты окисляются только очень сильными окислителями,
расщепляющими молекулу на кислоты и кетоны с меньшим числом углеродных
атомов.
Амины
можно рассматривать как
производные аммиака, получаемые последовательным замещением атомов водорода
алкильными группами, аналогично тому, как спирты и просто эфиры можно
представить как производные воды. В соответствии с числом замещенных атомов
водорода различают первичные (R–NH2),
вторичные (RRўNH)
и третичные амины (RRўRўўN).
Дальнейшее алкилирование дает четвертичные аммониевые соли RRўRўўRўўўN+X–,
которые можно рассматривать как
полностью алкилированную аммониевую соль NH4+X–.
Прямое
алкилирование аммиака алкилгалогенидами имеет очень ограниченную ценность
для получения первичных аминов, поскольку эту реакцию трудно контролировать
и она ведет к смесям первичных, вторичных и третичных продуктов. Прямое
алкилирование первичных аминов, однако, часто используют для получения
вторичных и третичных аминов, а также четвертичных аммониевых солей. Для
приготовления первичных аминов существует много хороших альтернативных
методов, например:
1)
расщепление амидов щелочным гипобромитом или гипохлоритом (реакция Гофмана).
Промежуточными соединениями в этой реакции являются изоцианаты, и этот
метод можно использовать для их получения:
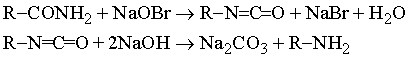
2)
перегруппировка азидов кислот в изоцианаты (см.
разд. IV-1.А.6)
с последующим их гидролизом до аминов (реакция Курциуса):

3)
реакция алкилгалогенидов с фталимидом калия с последующим гидролизом продукта
(реакция Габриэля):
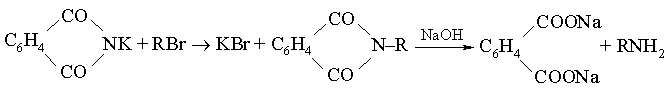
4)
восстановление нитрилов натрием в спирте или водородом на никелевом катализаторе
под давлением в присутствии аммиака:
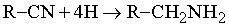
Другие
полезные методики этого рода включают восстановление нитросоединений R–NO2,
оксимов R–CH=NOH
и гидразонов R–CH=N–NH2.
Амины
– несколько более сильные основания, чем аммиак; константы диссоциации
для реакции  составляют примерно 10–4.
Поэтому амины легко образуют с большинством кислот соли, подобные солям
аммония. Первичные амины реагируют с азотистой кислотой, давая спирты
(
составляют примерно 10–4.
Поэтому амины легко образуют с большинством кислот соли, подобные солям
аммония. Первичные амины реагируют с азотистой кислотой, давая спирты
(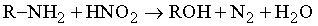 ),
но реакция является сложной и ведет также к олефинам и спиртам, изомерным
с ожидаемыми. При взаимодействии с нитрозилбромидом из аминов образуются
алкилбромиды ( ),
но реакция является сложной и ведет также к олефинам и спиртам, изомерным
с ожидаемыми. При взаимодействии с нитрозилбромидом из аминов образуются
алкилбромиды ( ).
Бензоилированные вторичные амины (R–NH–COC6H5)
при перегонке с пентабромидом
фосфора также превращаются в алкилбромиды (реакция Брауна). Вторичные
амины дают с азотистой кислотой N-нитрозопроизводные
RRўN–N=O,
тогда как третичные амины просто образуют соли. Это заметное различие
в поведении трех типов аминов по отношению к азотистой кислоте часто используется,
чтобы отличить их друг от друга. ).
Бензоилированные вторичные амины (R–NH–COC6H5)
при перегонке с пентабромидом
фосфора также превращаются в алкилбромиды (реакция Брауна). Вторичные
амины дают с азотистой кислотой N-нитрозопроизводные
RRўN–N=O,
тогда как третичные амины просто образуют соли. Это заметное различие
в поведении трех типов аминов по отношению к азотистой кислоте часто используется,
чтобы отличить их друг от друга.
Четвертичные аммониевые соли образуются
при действии алкилгалогенидов на третичные амины. Обработка оксидом серебра
превращает эти соли в соответствующие гидроксиды, которые являются основаниями,
сравнимыми по силе с гидроксидами натрия и калия. При нагревании они разлагаются
на амины и олефины с выделением воды. Такое поведение лежит в основе расщепления
по Гофману, широко использовавшегося при установлении структуры алкалоидов
для определения положения азота в цепи или гетероциклическом кольце. Амин
(например, 3-аминопентан) исчерпывающим метилированием (обработка избытком
метилиодида и сильным основанием) превращают в иодид триметилалкиламмония,
например (C2H5)2CH–N+(CH3)3I–,
и затем действием оксида серебра – в соответствующий гидроксид,
например (C2H5)2CH–N+(CH3)3OH–.
Последний при перегонке дает воду, триметиламин и олефин, например CH3CH=CHCH2CH3. Исходное положение аминогруппы можно вывести из положения двойной связи.
Первичные и вторичные (но не третичные)
амины реагируют с кислотами при нагревании, образуя амиды,
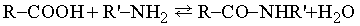
но
эта реакция обратима и чаще амиды получают из аминов и хлорангидридов
или ангидридов (см.
разд. IV-1.А.5).
С бензолсульфохлоридом первичные и вторичные амины дают бензолсульфамиды
(третичные амины опять-таки не реагируют по этой схеме):

Сульфамиды
первичных аминов растворимы в основаниях, а сульфамиды вторичных аминов
нейтральны; таким образом, эта реакция может быть использована для разделения
трех классов аминов (разделение по Гинзбергу).
Нитросоединения.
Эти соединения имеют
общую формулу R–NO2.
Их можно получать взаимодействием алкилгалогенидов с нитритом серебра
или натрия, а также взаимодействием углеводородов с азотной кислотой.
Вторичные и третичные водороды удается заместить нитрогруппами при нагревании
разветвленных углеводородов с концентрированной азотной кислотой в запаянной
трубке при 150°
С. Промышленное получение нитросоединений часто осуществляют в паровой
фазе при 400–500°
С. В промышленности нитросоединения используют как растворители и как
исходные соединения для получения алифатических аминов (см.
выше, методы получения первичных
аминов, п. 4).
Меркаптаны,
тиоэфиры и получаемые из них сернистые производные.
Меркаптаны (тиоспирты)
R–SH
свое название получили за легкость, с которой они образуют характерные
нерастворимые ртутные соли (corpus
mercurio aptum, т.е. тела, связывающие
ртуть). Меркаптаны получают действием гидросульфида калия на алкилгалогениды
или взаимодействием спиртов с сероводородом в паровой фазе при 300–350°
С над диоксидом тория. Они являются летучими жидкостями с неприятным запахом
и несколько более низкими температурами кипения, чем соответствующие спирты.
Подобно сероводороду, они представляют собой слабые кислоты (немного сильнее,
чем спирты) и легко образуют устойчивые металлические соли, или меркаптиды,
например R–SNa
или (R–S)2Pb.
Мягкие окислители, включая кислород воздуха, превращают их в диалкилдисульфиды
R–S–S–R,
из которых их можно регенерировать посредством самых различных восстановителей.
С альдегидами в присутствии хлороводорода или других кислых катализаторов
они дают меркаптаны, например RўCH(SR)2.
Тиоэфиры
R–S–Rў
можно получить взаимодействием алкилгалогенидов с сульфидом калия (2RI
+ K2S
® R–S–Rў
+ 2KI)
или с меркаптидами калия (RI
+ RўSK
® R–S–Rў
+ KI). Это летучие жидкости без
неприятного запаха, кипящие несколько выше, чем соответствующие меркаптаны.
С алкилгалогенидами они дают устойчивые кристаллические соли сульфония
RRўRўўS+X–,
которые по своим свойствами напоминают соли аммония. Сульфониевые основания
RRўRўўS+OH–
сравнимы по силе с едким натром. Окисление тиоэфиров через неусточивые
сульфоксиды RRўSO
ведет к стабильным сульфонам RRўSO2.
Окисление
меркаптанов под действием HNO3,
KMnO4
или H2O2
ведет в алкилсульфоновым кислотам R–SO3H.
Эти соединения можно также получить прямым сульфированием углеводородов
(RH + H2SO4
® RSO3H
+ H2O)
или взаимодействием сульфитов металлов с алкилгалогенидами. Они являются
сильными кислотами, легко растворимы в воде и дают кристаллические соли
(R–SO3M).
4.
С2+:
альдегиды и кетоны. Многие
альдегиды и кетоны, особенно альдегиды и кетоны терпенового ряда (терпены
– ненасыщенные углеводороды, получающиеся димеризацией изопрена
CH2=C(CH3)–CH=CH2),
встречаются в природе и имеют большое значение в парфюмерной промышленности.
Вещества этого рода включают цитронеллаль (CH3)2C=CHCH2CH2CH(CH3)CH2CHO
из масла цитронеллы и эвкалипта и цитраль (CH3)2C=CHCH2CH2C(CH3)=CHCHO
из масла цитронеллы.
Получение.
Формальдегид (т. кип. –21° С) получают каталитическим дегидрированием метанола над нагретым серебряным или медным катализатором. Водный 40%-й раствор формальдегида продается под названием формалин. Ацетальдегид получают каталитической гидратацией ацетилена. Ацетальдегид является исходным веществом для получения уксусной кислоты CH3COOH. Ацетон CH3COCH3 (пропанон), простейший кетон, представляет собой ценный растворитель. Его получают пропусканием паров уксусной кислоты над нагретым оксидом марганца:
 Общие методы получения альдегидов включают: а) окисление первичных спиртов хромовой или надсерной кислотами в присутствии иона двухвалентного железа, причем первый метод подходит только для получения летучих альдегидов, которые можно немедленно отогнать из реакционной смеси прежде, чем произойдет дальнейшее окисление; б) пиролиз кальциевой соли карбоновой кислоты с избытком муравьинокислого кальция:
Общие методы получения альдегидов включают: а) окисление первичных спиртов хромовой или надсерной кислотами в присутствии иона двухвалентного железа, причем первый метод подходит только для получения летучих альдегидов, которые можно немедленно отогнать из реакционной смеси прежде, чем произойдет дальнейшее окисление; б) пиролиз кальциевой соли карбоновой кислоты с избытком муравьинокислого кальция:
 и в) восстановление хлорангидридов RCOCl водородом над слегка дезактивированным палладием на сульфате бария (реакция Розенмунда).
Кетоны можно получить а) окислением вторичных спиртов хромовой кислотой при низкой температуре
и в) восстановление хлорангидридов RCOCl водородом над слегка дезактивированным палладием на сульфате бария (реакция Розенмунда).
Кетоны можно получить а) окислением вторичных спиртов хромовой кислотой при низкой температуре
 б) пиролизом кальциевых, бариевых и ториевых солей карбоновых кислот
б) пиролизом кальциевых, бариевых и ториевых солей карбоновых кислот
 в) действием алкилцинк- или алкилкадмийгалогенидов на ацилгалогениды
в) действием алкилцинк- или алкилкадмийгалогенидов на ацилгалогениды
 г) действием реактивов Гриньяра на нитрилы
г) действием реактивов Гриньяра на нитрилы
 Окисление
альдегидов до кислот лучше всего осуществлять при низкой температуре, используя а) перманганат калия KMnO4 в воде или ацетоне; б) надкислоты, например надуксусную кислоту CH3C(O)–O–OH, или в) аммиачный раствор нитрата серебра. Последний реагент полезен, если нужно отличить альдегиды от кетонов, поскольку небольшое количество альдегида вызывает образование красивого налета «серебряного зеркала» на стенках реакционного сосуда:
Окисление
альдегидов до кислот лучше всего осуществлять при низкой температуре, используя а) перманганат калия KMnO4 в воде или ацетоне; б) надкислоты, например надуксусную кислоту CH3C(O)–O–OH, или в) аммиачный раствор нитрата серебра. Последний реагент полезен, если нужно отличить альдегиды от кетонов, поскольку небольшое количество альдегида вызывает образование красивого налета «серебряного зеркала» на стенках реакционного сосуда:
 Окисление кетонов редко используется в синтетических целях. Особый тип окисления, известный как галоформная реакция, претерпевают ацетальдегид и метилкетоны (а также спирты, которые могут быть окислены в эти соединения) при действии гипогалогенитов (растворы галогенов в едком натре):
Окисление кетонов редко используется в синтетических целях. Особый тип окисления, известный как галоформная реакция, претерпевают ацетальдегид и метилкетоны (а также спирты, которые могут быть окислены в эти соединения) при действии гипогалогенитов (растворы галогенов в едком натре):
 Когда используется иод, образуется объемистый желтый осадок иодоформа CHI3. Эту реакцию применяют, чтобы отличить кетоны от их высших гомологов.
Восстановление
альдегидов в первичные спирты осуществляется каталитическим гидрированием при комнатной температуре над платиной в присутствии следов двухвалентного железа. Восстановления кетонов во вторичные спирты можно добиться подобным же образом над платиной, хромитом меди или никелем, хотя приходится использовать несколько более высокие давления водорода.
Когда используется иод, образуется объемистый желтый осадок иодоформа CHI3. Эту реакцию применяют, чтобы отличить кетоны от их высших гомологов.
Восстановление
альдегидов в первичные спирты осуществляется каталитическим гидрированием при комнатной температуре над платиной в присутствии следов двухвалентного железа. Восстановления кетонов во вторичные спирты можно добиться подобным же образом над платиной, хромитом меди или никелем, хотя приходится использовать несколько более высокие давления водорода.
Альдегиды и кетоны можно восстановить в спирты литийалюминийгидридом LiAlH4 или борогидридом натрия NaBH4. Карбонильную группу можно восстановить до метиленовой по Клемменсену (амальгамой цинка в соляной кислоте) или по Кижнеру – Вольфу (обработка гидразона горячей щелочью).
Полимеризация.
Альдегиды склонны полимеризоваться, особенно при контакте с воздухом или в присутствии кислотных либо основных катализаторов. При кислотном катализе полимеризация формальдегида ведет к образованию тримера – триоксана (или a-триоксиметилена)
 а также к полимерам с прямой цепью различной длины, называемым полиоксиметиленами, HO–CH2–(OCH2)n–O–CH2OH, содержащим до 100 формальдегидных единиц. Триоксан представляет собой удобную твердую модификацию формальдегида, поскольку из него можно регенерировать газообразный мономер просто нагреванием. В присутствии гидроксида кальция формальдегид может давать смесь оксиальдегидов и оксикетонов общей формулы (CH2O)n, из которой были выделены вещества, напоминающие сахара. Аналогичным образом ацетальдегид претерпевает кислотно-катализируемую тримеризацию в паральдегид
а также к полимерам с прямой цепью различной длины, называемым полиоксиметиленами, HO–CH2–(OCH2)n–O–CH2OH, содержащим до 100 формальдегидных единиц. Триоксан представляет собой удобную твердую модификацию формальдегида, поскольку из него можно регенерировать газообразный мономер просто нагреванием. В присутствии гидроксида кальция формальдегид может давать смесь оксиальдегидов и оксикетонов общей формулы (CH2O)n, из которой были выделены вещества, напоминающие сахара. Аналогичным образом ацетальдегид претерпевает кислотно-катализируемую тримеризацию в паральдегид
 а при низких температурах – тетрамеризацию в метальдегид.
Альдольная конденсация
– это реакция димеризации, которую претерпевают альдегиды и (несколько труднее) кетоны, имеющие водород при углеводородном атоме, соседнем с карбонильной группой. Так, слабые основания вызывают обратимую димеризацию ацетальдегида в альдоль, CH3–CHOH–CH2CHO, а ацетона – в диацетоновый спирт (CH3)2C(OH)CH2COCH3. Кислоты вызывают такую же конденсацию, но далее катализируют отщепление воды, ведущее к ненасыщенным альдегидам и кетонам, которые в свою очередь могут претерпевать дальнейшую конденсацию:
а при низких температурах – тетрамеризацию в метальдегид.
Альдольная конденсация
– это реакция димеризации, которую претерпевают альдегиды и (несколько труднее) кетоны, имеющие водород при углеводородном атоме, соседнем с карбонильной группой. Так, слабые основания вызывают обратимую димеризацию ацетальдегида в альдоль, CH3–CHOH–CH2CHO, а ацетона – в диацетоновый спирт (CH3)2C(OH)CH2COCH3. Кислоты вызывают такую же конденсацию, но далее катализируют отщепление воды, ведущее к ненасыщенным альдегидам и кетонам, которые в свою очередь могут претерпевать дальнейшую конденсацию:
 Реакции присоединения по карбонилу.
Карбонильная группа является ненасыщенной и потому вступает в многочисленные реакции присоединения. Так (см. разд. IV-1.А.1), получение спиртов легко осуществляется присоединением реактивов Гриньяра по карбонильной группе. Присоединение синильной кислоты ведет к образованию циангидринов (реакция Килиани)
Реакции присоединения по карбонилу.
Карбонильная группа является ненасыщенной и потому вступает в многочисленные реакции присоединения. Так (см. разд. IV-1.А.1), получение спиртов легко осуществляется присоединением реактивов Гриньяра по карбонильной группе. Присоединение синильной кислоты ведет к образованию циангидринов (реакция Килиани)  , а в присутствии хлорида аммония – аминонитрилов R–CH(NH2)–CN. Большое число азотистых оснований общего типа A–NH2 дает при присоединении к карбонильной группе с последующим отщеплением воды кристаллические производные типа A–N=CHR и A–N=CRRў. Так, гидроксиламин NH2OH дает оксимы RCH=NOH; гидразин NH2–NH2 – гидразоны RCH=N–NH2, а семикарбазид H2N–NH–CO–NH2 – семикарбазоны RCH=N–NHCONH2, вещества, ценные для очистки и идентификации карбонильных соединений. Бисульфит натрия присоединяется аналогичным образом к альдегидам и метилкетонам, давая кристаллические водорастворимые производные R–CH(OH)–SO3Na, из которых карбонильные соединения можно регенерировать действием карбоната натрия.
Ацетали.
Альдегиды легко реагируют со спиртами в присутствии кислотных катализаторов, образуя ацетали: , а в присутствии хлорида аммония – аминонитрилов R–CH(NH2)–CN. Большое число азотистых оснований общего типа A–NH2 дает при присоединении к карбонильной группе с последующим отщеплением воды кристаллические производные типа A–N=CHR и A–N=CRRў. Так, гидроксиламин NH2OH дает оксимы RCH=NOH; гидразин NH2–NH2 – гидразоны RCH=N–NH2, а семикарбазид H2N–NH–CO–NH2 – семикарбазоны RCH=N–NHCONH2, вещества, ценные для очистки и идентификации карбонильных соединений. Бисульфит натрия присоединяется аналогичным образом к альдегидам и метилкетонам, давая кристаллические водорастворимые производные R–CH(OH)–SO3Na, из которых карбонильные соединения можно регенерировать действием карбоната натрия.
Ацетали.
Альдегиды легко реагируют со спиртами в присутствии кислотных катализаторов, образуя ацетали:
 Реакция обратима, альдегиды и спирты легко регенерировать из ацеталей действием воды и кислот; образование ацеталей часто используют, чтобы временно защитить чувствительную альдегидную группу, пока в другой части молекулы выполняются химические превращения. Кетоны обычно не столь легко вступают в эту реакцию, но ацетон конденсируется с полигидроксисоединениями (например, сахарами), образуя циклические ацетали типа
Реакция обратима, альдегиды и спирты легко регенерировать из ацеталей действием воды и кислот; образование ацеталей часто используют, чтобы временно защитить чувствительную альдегидную группу, пока в другой части молекулы выполняются химические превращения. Кетоны обычно не столь легко вступают в эту реакцию, но ацетон конденсируется с полигидроксисоединениями (например, сахарами), образуя циклические ацетали типа
 Галогенирование.
Карбонильные соединения, имеющие водород при углероде, соседнем с карбонильной группой, легко реагируют с галогенами, давая a-галогенальдегиды или кетоны. Реакция катализируется кислотами и основаниями. Кислотно-катализируемое галогенирование несколько легче контролировать, и потому его часто используют, если желательно ввести только один атом галогена:
Галогенирование.
Карбонильные соединения, имеющие водород при углероде, соседнем с карбонильной группой, легко реагируют с галогенами, давая a-галогенальдегиды или кетоны. Реакция катализируется кислотами и основаниями. Кислотно-катализируемое галогенирование несколько легче контролировать, и потому его часто используют, если желательно ввести только один атом галогена:
 Атом галогена, соседний с карбонильной группой, исключительно активен во многих реакциях замещения. Часто его можно также отщепить вместе с соседним водородом при действии оснований, что дает ненасыщенные карбонильные соединения R–C(O)–CH=CHRў.
5. С3+: карбоновые кислоты и их производные.
Нахождение в природе.
В природе карбоновые кислоты редко находятся в свободном виде, хотя ожоги крапивой и укусы муравьев и пчел болезненны из-за раздражающего действия свободной муравьиной кислоты. Среди других карбоновых кислот, встречающихся в природе в свободном виде, – изовалериановая (корень валерианы) и изомасляная. В природе карбоновые кислоты встречаются главным образом в виде эфиров и соединений со спиртами. Первые три члена гомологического ряда карбоновых кислот являются относительно высококипящими жидкостями с резким, раздражающим запахом. Высшие члены ряда представляют собой жидкости или низкоплавкие твердые вещества, причем более летучие кислоты обладают крайне неприятным запахом.
Получение.
В промышленности натриевую соль муравьиной кислоты получают взаимодействием монооксида углерода с едким натром при 200° С под давлением. При осторожном нагревании этой соли с рассчитанным количеством серной кислоты удается отогнать муравьиную кислоту. Уксусную кислоту получают в промышленных масштабах главным образом окислением уксусного альдегида, получаемого из ацетилена. Водный 5% раствор (столовый уксус) можно приготовить разбавлением промышленной уксусной кислоты или сбраживанием разбавленных растворов спирта уксуснокислыми бактериями.
Атом галогена, соседний с карбонильной группой, исключительно активен во многих реакциях замещения. Часто его можно также отщепить вместе с соседним водородом при действии оснований, что дает ненасыщенные карбонильные соединения R–C(O)–CH=CHRў.
5. С3+: карбоновые кислоты и их производные.
Нахождение в природе.
В природе карбоновые кислоты редко находятся в свободном виде, хотя ожоги крапивой и укусы муравьев и пчел болезненны из-за раздражающего действия свободной муравьиной кислоты. Среди других карбоновых кислот, встречающихся в природе в свободном виде, – изовалериановая (корень валерианы) и изомасляная. В природе карбоновые кислоты встречаются главным образом в виде эфиров и соединений со спиртами. Первые три члена гомологического ряда карбоновых кислот являются относительно высококипящими жидкостями с резким, раздражающим запахом. Высшие члены ряда представляют собой жидкости или низкоплавкие твердые вещества, причем более летучие кислоты обладают крайне неприятным запахом.
Получение.
В промышленности натриевую соль муравьиной кислоты получают взаимодействием монооксида углерода с едким натром при 200° С под давлением. При осторожном нагревании этой соли с рассчитанным количеством серной кислоты удается отогнать муравьиную кислоту. Уксусную кислоту получают в промышленных масштабах главным образом окислением уксусного альдегида, получаемого из ацетилена. Водный 5% раствор (столовый уксус) можно приготовить разбавлением промышленной уксусной кислоты или сбраживанием разбавленных растворов спирта уксуснокислыми бактериями.
Общие методы получения кислот в лаборатории:
а) окисление первичных спиртов азотной кислотой, перманганатом калия или хромовой кислотой  ; ;
б) обработка реактивов Гриньяра углекислым газом  ; ;
в) взаимодействие алкилгалогенидов с цианидом натрия с последующим гидролизом образующихся нитрилов  ; ;  ; ;
г) окислительное расщепление олефинов перманганатом калия  .
В неорганической химии имеется правило, согласно которому окисление элемента, несущего гидроксильную группу, усиливает кислотные свойства этой группы (ср. H2SO4 и H2SO3, HNO3 и HNO2). Аналогично в органической химии карбоновые кислоты проявляют значительно более кислотные свойства, чем спирты (RCOOH и RCH2OH). Константы диссоциации для равновесия RCOOH .
В неорганической химии имеется правило, согласно которому окисление элемента, несущего гидроксильную группу, усиливает кислотные свойства этой группы (ср. H2SO4 и H2SO3, HNO3 и HNO2). Аналогично в органической химии карбоновые кислоты проявляют значительно более кислотные свойства, чем спирты (RCOOH и RCH2OH). Константы диссоциации для равновесия RCOOH RCOO– + H+ обычно имеют величину 10–5–10–4 (для спиртов – 10–18). Ввиду достаточно высокой кислотности карбоновых кислот они легко растворяются в бикарбонате натрия, соде или едком натре с образованием натриевых солей (RCOONa). Из них карбоновые кислоты можно выделить действием более сильных минеральных кислот (HCl, H2SO4).
Со спиртами карбоновые кислоты реагируют, давая сложные эфиры (RCOOH + RўOHRCOORў + H2O). Прямая реакция идет медленно, но катализируется сильными неорганическими кислотами. Она обратима, поэтому успешное приготовление сложного эфира из кислоты может быть достигнуто только смещением равновесия путем использования большого избытка спирта или удаления воды из реакционной смеси. RCOO– + H+ обычно имеют величину 10–5–10–4 (для спиртов – 10–18). Ввиду достаточно высокой кислотности карбоновых кислот они легко растворяются в бикарбонате натрия, соде или едком натре с образованием натриевых солей (RCOONa). Из них карбоновые кислоты можно выделить действием более сильных минеральных кислот (HCl, H2SO4).
Со спиртами карбоновые кислоты реагируют, давая сложные эфиры (RCOOH + RўOHRCOORў + H2O). Прямая реакция идет медленно, но катализируется сильными неорганическими кислотами. Она обратима, поэтому успешное приготовление сложного эфира из кислоты может быть достигнуто только смещением равновесия путем использования большого избытка спирта или удаления воды из реакционной смеси.
Из кислоты можно получить метиловые эфиры в очень мягких условиях действием диазометана:
 Карбоновые кислоты реагируют с трихлоридом фосфора, пентахлоридом фосфора или тионилхлоридом (SOCl2) с образованием высокореакционноспособных хлорангидридов RCOCl:
Карбоновые кислоты реагируют с трихлоридом фосфора, пентахлоридом фосфора или тионилхлоридом (SOCl2) с образованием высокореакционноспособных хлорангидридов RCOCl:
 Эти очень легко гидролизуемые вещества особенно полезны для получения производных кислот. Так, со спиртами они дают эфиры:
Эти очень легко гидролизуемые вещества особенно полезны для получения производных кислот. Так, со спиртами они дают эфиры:
 с аммиаком или аминами – амиды:
с аммиаком или аминами – амиды:
 с натриевыми солями карбоновых кислот – ангидриды:
с натриевыми солями карбоновых кислот – ангидриды:
 Соли кислот вступают в некоторые полезные реакции. Так, натриевые соли при сплавлении с едким натром дают насыщенные углеводороды RH. При электролизе получаются углеводороды RR. Перегонка кальциевых или ториевых солей приводит к образованию кетонов RCOR. Серебряные соли RCOOAg при обработке алкилгалогенидами RўBr дают эфиры RCOORў, а при действии галогенов – алкилгалогениды:
Соли кислот вступают в некоторые полезные реакции. Так, натриевые соли при сплавлении с едким натром дают насыщенные углеводороды RH. При электролизе получаются углеводороды RR. Перегонка кальциевых или ториевых солей приводит к образованию кетонов RCOR. Серебряные соли RCOOAg при обработке алкилгалогенидами RўBr дают эфиры RCOORў, а при действии галогенов – алкилгалогениды:  .
Эфиры: жиры и воски.
Эти соединения, препаративные методы получения которых обсуждены выше, можно рассматривать как образующиеся из кислоты и спирта при отщеплении воды: .
Эфиры: жиры и воски.
Эти соединения, препаративные методы получения которых обсуждены выше, можно рассматривать как образующиеся из кислоты и спирта при отщеплении воды:  . Летучие эфиры, имеющие низкие молекулярные массы, являются душистыми маслами. Многие из них, например этилформиат, этилацетат, изоамилацетат, этилбутират и т.д., обнаружены в душистых цветах и плодах. Наиболее важными среди эфиров с высокой молекулярной массой являются жиры и воски, которые представляют собой масла или низкоплавкие твердые вещества. . Летучие эфиры, имеющие низкие молекулярные массы, являются душистыми маслами. Многие из них, например этилформиат, этилацетат, изоамилацетат, этилбутират и т.д., обнаружены в душистых цветах и плодах. Наиболее важными среди эфиров с высокой молекулярной массой являются жиры и воски, которые представляют собой масла или низкоплавкие твердые вещества.
Жиры – это триэфиры глицерина ( см. ниже) и высших жирных кислот:
 Жирные кислоты из природных жиров содержат четное число углеродных атомов в молекуле, причем самыми распространенными являются С16- и С18-кислоты. Кроме насыщенных кислот, в жирах присутствуют многочисленные ненасыщенные кислоты. Высоконенасыщенные жиры – жидкости (растительные и рыбные жиры), тогда как более насыщенные жиры (жиры животных) – твердые вещества. Твердые кулинарные жиры можно получить из жидких жиров частичным гидрированием водородом на никелевом катализаторе. Наиболее обычными из ненасыщенных кислот являются
Жирные кислоты из природных жиров содержат четное число углеродных атомов в молекуле, причем самыми распространенными являются С16- и С18-кислоты. Кроме насыщенных кислот, в жирах присутствуют многочисленные ненасыщенные кислоты. Высоконенасыщенные жиры – жидкости (растительные и рыбные жиры), тогда как более насыщенные жиры (жиры животных) – твердые вещества. Твердые кулинарные жиры можно получить из жидких жиров частичным гидрированием водородом на никелевом катализаторе. Наиболее обычными из ненасыщенных кислот являются
 Глицериды, как и все эфиры, гидролизуются (омыляются) водными щелочами с образованием спирта (глицерина) и соли щелочных металлов входящих в их состав кислот. Соли высших кислот называются мылами. Процесс омыления имеет большое промышленное значение. Во время войн глицерин пользовался большим спросом для получения взрывчатых веществ (см. разд. IV-1.Б.1).
Воски являются сложными эфирами высших жирных кислот и высших спиртов. Примерами могут служить пчелиный воск (мирицилпальмитат C15H31COOC31H63) и карнаубский воск (церилцеротат C25H51COOC26H53).
Кроме реакций омыления, обсуждавшихся выше, эфиры вступают в общие для них реакции сольволиза с водой, спиртами или аммиаком:
Глицериды, как и все эфиры, гидролизуются (омыляются) водными щелочами с образованием спирта (глицерина) и соли щелочных металлов входящих в их состав кислот. Соли высших кислот называются мылами. Процесс омыления имеет большое промышленное значение. Во время войн глицерин пользовался большим спросом для получения взрывчатых веществ (см. разд. IV-1.Б.1).
Воски являются сложными эфирами высших жирных кислот и высших спиртов. Примерами могут служить пчелиный воск (мирицилпальмитат C15H31COOC31H63) и карнаубский воск (церилцеротат C25H51COOC26H53).
Кроме реакций омыления, обсуждавшихся выше, эфиры вступают в общие для них реакции сольволиза с водой, спиртами или аммиаком:
 Первые две реакции катализируются неорганическими кислотами. Таким образом, данный сложный эфир при правильном выборе сольволитического агента можно превратить в свободную кислоту, другой эфир или амид.
Некоторое препаративное значение имеет реакция восстановления сложных эфиров в спирты. Это может быть осуществлено действием натрия в спирте или гидрированием под давлением при 250° С в присутствии хромита меди. Последний метод используется в промышленности для получения многих малодоступных ранее высших спиртов.
Первые две реакции катализируются неорганическими кислотами. Таким образом, данный сложный эфир при правильном выборе сольволитического агента можно превратить в свободную кислоту, другой эфир или амид.
Некоторое препаративное значение имеет реакция восстановления сложных эфиров в спирты. Это может быть осуществлено действием натрия в спирте или гидрированием под давлением при 250° С в присутствии хромита меди. Последний метод используется в промышленности для получения многих малодоступных ранее высших спиртов.
Ортоэфиры RC(ORў)3, как и ацетали, вполне устойчивы в щелочных растворах, но легко превращаются в сложные эфиры при действии водных кислот. Трихлорметильные соединения RCCl3 можно прогидролизовать до кислот нагреванием с водной щелочью.
Сложные эфиры конденсируются в b-кетоэфиры в присутствии этилата натрия C2H5ONa или сходных с ним сильных оснований (см. разд. IV-1.Б.5).
Другие производные кислот: амиды и ангидриды.
Амиды RCONH2 получают в основном действием аммиака на хлорангидриды RCOCl или на сложные эфиры RCOORў. Они имеют значение в органическом синтезе, поскольку из них можно приготовить первичные амины RNH2 по реакции Гофмана (разд. IV-1.А.3). При дегидратации фосфорным ангидридом из них образуются нитрилы R–CєN.
Ангидриды RC(O)OC(O)R обычно получают действием хлорангидридов на натриевые соли кислот. Они используются в основном для ацилирования спиртов. Так, действием уксусного ангидрида на спирты получают ацетаты:
 Кислоты, сложные эфиры, хлорангидриды и ангидриды можно восстановить до спиртов литийалюминийгидридом.
6. С4+: производные угольной кислоты.
Эфиры угольной и хлоругольной кислот.
При взаимодействии фосгена COCl2 со спиртами первоначально образуются эфиры хлоругольной кислоты R–O–COCl. Эти вещества представляют собой летучие жидкости с острым запахом, применяемые для введения этерифицированной карбоксильной группы в спирты, амины и другие соединения, имеющие активный замещаемый водород. Дальнейшее взаимодействие хлоркарбонатов со спиртами дает карбонаты RO–CO–ORў – летучие жидкости с общими для эфиров свойствами.
Ксантаты.
Взаимодействие сероуглерода со спиртами в присутствии щелочей ведет к образованию ксантатов – сложноэфирных солей эфиров дитиоугольной кислоты:
Кислоты, сложные эфиры, хлорангидриды и ангидриды можно восстановить до спиртов литийалюминийгидридом.
6. С4+: производные угольной кислоты.
Эфиры угольной и хлоругольной кислот.
При взаимодействии фосгена COCl2 со спиртами первоначально образуются эфиры хлоругольной кислоты R–O–COCl. Эти вещества представляют собой летучие жидкости с острым запахом, применяемые для введения этерифицированной карбоксильной группы в спирты, амины и другие соединения, имеющие активный замещаемый водород. Дальнейшее взаимодействие хлоркарбонатов со спиртами дает карбонаты RO–CO–ORў – летучие жидкости с общими для эфиров свойствами.
Ксантаты.
Взаимодействие сероуглерода со спиртами в присутствии щелочей ведет к образованию ксантатов – сложноэфирных солей эфиров дитиоугольной кислоты:
 Это кристаллические вещества, которые при обработке кислотами снова выделяют спирты. Применение этой реакции к целлюлозе приводит к получению вискозы – коллоидного раствора ксантата целлюлозы
Это кристаллические вещества, которые при обработке кислотами снова выделяют спирты. Применение этой реакции к целлюлозе приводит к получению вискозы – коллоидного раствора ксантата целлюлозы
 который при выдавливании в кислый раствор дает красивые нити регенерированной целлюлозы или вискозного шелка.
Производные карбаминовой кислоты.
Карбаминовая (моноамидугольная) кислота H2NCOOH известна только в форме солей, сложных эфиров и амидов. Ее эфиры H2NCOOR (уретаны) – кристаллические вещества. Многие из них используются в медицине как седативные средства, например апонал H2NCOOC(CH3)2C2H5 и гедонал H2NCOOCH(CH3)C3H7. Они могут быть получены взаимодействием а) аммиака и эфиров хлоругольной кислоты, б) карбамилхлорида ClCONH2 и спиртов, в) мочевины и спиртов. Замещенные по азоту уретаны получают действием спиртов на изоцианаты
который при выдавливании в кислый раствор дает красивые нити регенерированной целлюлозы или вискозного шелка.
Производные карбаминовой кислоты.
Карбаминовая (моноамидугольная) кислота H2NCOOH известна только в форме солей, сложных эфиров и амидов. Ее эфиры H2NCOOR (уретаны) – кристаллические вещества. Многие из них используются в медицине как седативные средства, например апонал H2NCOOC(CH3)2C2H5 и гедонал H2NCOOCH(CH3)C3H7. Они могут быть получены взаимодействием а) аммиака и эфиров хлоругольной кислоты, б) карбамилхлорида ClCONH2 и спиртов, в) мочевины и спиртов. Замещенные по азоту уретаны получают действием спиртов на изоцианаты
 Изоцианаты получают по реакции Гофмана или Курциуса.
Мочевину NH2CONH2 можно рассматривать как диамид угольной кислоты или амид карбаминовой кислоты. У млекопитающих она выделяется как главный конечный продукт азотного обмена. Взрослый человек выделяет в день 20–30 г мочевины. Историческое значение мочевины состоит в том, что ее синтезом из цианата аммония Вёлер в 1828 показал отсутствие мистического барьера между органической и неорганической химией. Синтез Вёлера еще используется как источник этого соединения:
Изоцианаты получают по реакции Гофмана или Курциуса.
Мочевину NH2CONH2 можно рассматривать как диамид угольной кислоты или амид карбаминовой кислоты. У млекопитающих она выделяется как главный конечный продукт азотного обмена. Взрослый человек выделяет в день 20–30 г мочевины. Историческое значение мочевины состоит в том, что ее синтезом из цианата аммония Вёлер в 1828 показал отсутствие мистического барьера между органической и неорганической химией. Синтез Вёлера еще используется как источник этого соединения:
 хотя более новый процесс, состоящий в нагревании карбамата аммония под давлением, уже заменил его в промышленности:
хотя более новый процесс, состоящий в нагревании карбамата аммония под давлением, уже заменил его в промышленности:
 Мочевину получают также взаимодействием цианамида NH2CN с водой. Замещенные мочевины можно синтезировать действием аминов на фосген или изоцианаты:
Мочевину получают также взаимодействием цианамида NH2CN с водой. Замещенные мочевины можно синтезировать действием аминов на фосген или изоцианаты:
 Мочевина имеет промышленное значение как удобрение и как промежуточное вещество в получении снотворных и успокоительных средств типа
Мочевина имеет промышленное значение как удобрение и как промежуточное вещество в получении снотворных и успокоительных средств типа
 например веронала (R = Rў = C2H5), люминала (R = C6H5, Rў = C2H5) и пропанала (R = Rў = C3H7).
Гуанидин HN=C(NH2)2 – имидопроизводное мочевины; его тиоцианат можно получить нагреванием тиоцианата аммония NH4SCH до 150° С. Гуанидин образуется также при взаимодействии аммиака с цианамидом. Это бесцветное кристаллическое гигроскопичное вещество с сильными основными свойствами.
например веронала (R = Rў = C2H5), люминала (R = C6H5, Rў = C2H5) и пропанала (R = Rў = C3H7).
Гуанидин HN=C(NH2)2 – имидопроизводное мочевины; его тиоцианат можно получить нагреванием тиоцианата аммония NH4SCH до 150° С. Гуанидин образуется также при взаимодействии аммиака с цианамидом. Это бесцветное кристаллическое гигроскопичное вещество с сильными основными свойствами.
Тиомочевина S=C(NH2)2 – сернистый аналог мочевины – может быть получена перегруппировкой тиоцианата аммония в его точке плавления, 149° С. Тиомочевина легко реагирует с алкилгалогенидами, давая кристаллические алкилтиурониевые соли [RS–(NH2)C=NH2+]X–.
Производные циановой и тиоциановой кислот.
Алкилизоцианаты R–N=C=O – промежуточные продукты в реакции Гофмана (превращение амидов карбоновых кислот в первичные амины под действием гипогалогенитов щелочных металлов) и реакции Курциуса (получение первичных аминов термической перегруппировкой ацилазидов в изоцианаты с последующим их гидролизом). Их можно также получить взаимодействием гидрохлоридов аминов (RNH3Cl) и фосгена. Это летучие жидкости с острым запахом, которые легко тримеризуются в циануровую кислоту
 При взаимодействии со спиртами они дают уретаны (см. выше), а при кислотном или щелочном гидролизе – амины.
Сернистые аналоги этих соединений – изотиоцианаты – летучие жидкости, известные как горчичные масла (это название связано с их острым запахом). Многие из них встречаются в природе в виде гликозидов (см. ГЛИКОЗИДЫ). Так, аллилгорчичное масло CH2=CHCH2N=C=S находится в виде гликозида в черной горчице; этот гликозид может быть расщеплен под действием кислот или фермента мирозина:
При взаимодействии со спиртами они дают уретаны (см. выше), а при кислотном или щелочном гидролизе – амины.
Сернистые аналоги этих соединений – изотиоцианаты – летучие жидкости, известные как горчичные масла (это название связано с их острым запахом). Многие из них встречаются в природе в виде гликозидов (см. ГЛИКОЗИДЫ). Так, аллилгорчичное масло CH2=CHCH2N=C=S находится в виде гликозида в черной горчице; этот гликозид может быть расщеплен под действием кислот или фермента мирозина:
 Б. ПОЛИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ
В органической химии существуют неограниченные возможности для получения соединений, имеющих несколько различных функциональных групп (например, –OH, –COOH, –C(=O)–) в одной и той же молекуле. В общем случае, если такие группы достаточно удалены друг от друга, можно считать, что они будут обнаруживать поведение и реакции, характерные для каждой из них в отдельности. Однако, если они расположены близко друг к другу, возникают классы соединений, требующие особого рассмотрения. Ниже приведены некоторые наиболее важные примеры.
1. Многоатомные спирты и их производные.
Гликоли.
Простейшим многоатомным спиртом является двухатомный (т.е. две OH-группы) вицинальный (т.е. OH-группы находятся при соседних атомах С) спирт этиленгликоль CH2OH–CH2OH (этандиол), который может быть получен из этиленхлоргидрина CH2OH–CH2Cl гидролизом в присутствии карбоната натрия. Этиленгликоль находит широкое использование как антифриз, а его моноалкиловые эфиры RO–CH2CH2OH, известные под названием целлозольвы, широко применяются как растворители для полиролей и лаков. Высшие гомологи этиленгликоля включают гликоли R–CHOH–CHOH–Rў, семипинаконы (RRўCOH–CHOH–Rўў) и пинаконы (RRўCOH–COHRўўRўўў). Общие методы их получения включают гидролиз соответствующих вицинальных дигалогенидов (RCHBr–CHBrRў) или галогенгидринов (RCHOH–CHBrRў), которые образуются (см. разд. IV-1.А.2) присоединением галогенов или гипогалогеновых кислот (HOX) к олефинам; олефиновая двойная связь может быть также непосредственно окислена холодным разбавленным перманганатом калия, тетраоксидом осмия, пероксидом водорода или его ацилпроизводными, например надуксусной кислотой CH3–C(O)–O–OH.
В дополнение к обычным реакциям спиртов гликоли особенно чувствительны к окислительному расщеплению. Так, они количественно окисляются до альдегидов иодной кислотой или тетраацетатом свинца:
Б. ПОЛИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ
В органической химии существуют неограниченные возможности для получения соединений, имеющих несколько различных функциональных групп (например, –OH, –COOH, –C(=O)–) в одной и той же молекуле. В общем случае, если такие группы достаточно удалены друг от друга, можно считать, что они будут обнаруживать поведение и реакции, характерные для каждой из них в отдельности. Однако, если они расположены близко друг к другу, возникают классы соединений, требующие особого рассмотрения. Ниже приведены некоторые наиболее важные примеры.
1. Многоатомные спирты и их производные.
Гликоли.
Простейшим многоатомным спиртом является двухатомный (т.е. две OH-группы) вицинальный (т.е. OH-группы находятся при соседних атомах С) спирт этиленгликоль CH2OH–CH2OH (этандиол), который может быть получен из этиленхлоргидрина CH2OH–CH2Cl гидролизом в присутствии карбоната натрия. Этиленгликоль находит широкое использование как антифриз, а его моноалкиловые эфиры RO–CH2CH2OH, известные под названием целлозольвы, широко применяются как растворители для полиролей и лаков. Высшие гомологи этиленгликоля включают гликоли R–CHOH–CHOH–Rў, семипинаконы (RRўCOH–CHOH–Rўў) и пинаконы (RRўCOH–COHRўўRўўў). Общие методы их получения включают гидролиз соответствующих вицинальных дигалогенидов (RCHBr–CHBrRў) или галогенгидринов (RCHOH–CHBrRў), которые образуются (см. разд. IV-1.А.2) присоединением галогенов или гипогалогеновых кислот (HOX) к олефинам; олефиновая двойная связь может быть также непосредственно окислена холодным разбавленным перманганатом калия, тетраоксидом осмия, пероксидом водорода или его ацилпроизводными, например надуксусной кислотой CH3–C(O)–O–OH.
В дополнение к обычным реакциям спиртов гликоли особенно чувствительны к окислительному расщеплению. Так, они количественно окисляются до альдегидов иодной кислотой или тетраацетатом свинца:
 Семипинаконы и пинаконы расщепляются аналогичным образом. Пинаконы особенно чувствительны к действию неорганических кислот, в присутствии которых происходит их молекулярная перегруппировка в пинаколины:
Семипинаконы и пинаконы расщепляются аналогичным образом. Пинаконы особенно чувствительны к действию неорганических кислот, в присутствии которых происходит их молекулярная перегруппировка в пинаколины:
 Глицерин
CH2OH–CHOH–CH2OH – простейший трехатомный спирт, промежуточный продукт в производстве мыла, образуется при омылении жиров (см. разд. IV-1.А.5). Его триэфир азотной кислоты CH2NO3–CHNO3–CH2NO3 – взрывчатое вещество нитроглицерин – образуется при обработке глицерина смесью азотной и серной кислот. Нитроглицерин взрывается от малейшего сотрясения, но адсорбция на кизельгуре (диатомит) повышает его устойчивость; в таком виде он известен как динамит.
Глицерин
CH2OH–CHOH–CH2OH – простейший трехатомный спирт, промежуточный продукт в производстве мыла, образуется при омылении жиров (см. разд. IV-1.А.5). Его триэфир азотной кислоты CH2NO3–CHNO3–CH2NO3 – взрывчатое вещество нитроглицерин – образуется при обработке глицерина смесью азотной и серной кислот. Нитроглицерин взрывается от малейшего сотрясения, но адсорбция на кизельгуре (диатомит) повышает его устойчивость; в таком виде он известен как динамит.
Трехатомные спирты, как и гликоли, подвержены окислительному расщеплению иодной кислотой:
 2. Гидроксиальдегиды и кетоны.
a-Гидроксикарбонильные соединения
2. Гидроксиальдегиды и кетоны.
a-Гидроксикарбонильные соединения
 могут быть получены действием бикарбоната натрия на соответствующие a-галогеноангидриды и кетоны могут быть получены действием бикарбоната натрия на соответствующие a-галогеноангидриды и кетоны  . a-Кетоспирты типа R–CO–CH2OH получаются кислотно-катализируемым гидролизом диазокетонов R–CO–CHN2, которые могут быть получены действием диазометана CH2N2 на ацилхлориды (хлорангидриды карбоновых кислот). При взаимодействии a-кетоспиртов с тетраацетатом свинца образуются их ацетаты: . a-Кетоспирты типа R–CO–CH2OH получаются кислотно-катализируемым гидролизом диазокетонов R–CO–CHN2, которые могут быть получены действием диазометана CH2N2 на ацилхлориды (хлорангидриды карбоновых кислот). При взаимодействии a-кетоспиртов с тетраацетатом свинца образуются их ацетаты:
 a-Гидроксикарбонильная группировка часто встречается в природных продуктах, она свойственна сахарам, например альдозам HOCH2–(CHOH)n–CHO (n = 2 – 4) и кетозам CH2OH–(CHOH)n–CO–CH2OH (n = 2, 3). Она легко окисляется; так, под действием иодной кислоты и тетрацетата свинца a-кетоспирты подвергаются мягкому расщеплению:
a-Гидроксикарбонильная группировка часто встречается в природных продуктах, она свойственна сахарам, например альдозам HOCH2–(CHOH)n–CHO (n = 2 – 4) и кетозам CH2OH–(CHOH)n–CO–CH2OH (n = 2, 3). Она легко окисляется; так, под действием иодной кислоты и тетрацетата свинца a-кетоспирты подвергаются мягкому расщеплению:
 В концевом положении, например –CHOH–CHO или –COCH2OH, группировка взаимодействует с аммиачным раствором нитрата серебра, давая серебряное зеркало, и с ионом меди в щелочном растворе тартрата (фелингова жидкость) или цитрата (реактив Бенедикта), образуя красный осадок оксида меди(I), или медное зеркало, а также с фенилгидразином C6H5NHNH2, давая озазоны (C6H5)HNN=C(R)–CH=N–NHC6H5, которые можно гидролизовать в соответствующие a-кетоальдегиды (дикарбонильные соединения).
b-Гидроксикарбонильные соединения.
Когда ацетальдегид обрабатывают мягким основанием (например, карбонатом калия), происходит его димеризация в альдоль (b-гидроксиальдегид) CH3CHOHCH2CHO. Аналогично ацетон может димеризоваться в диацетоновой спирт (CH3)2C(OH)CH2C(O)CH3 (b-гидроксикетон). Эта реакция представляет собой метод получения b-гидроксикарбонильных соединений, или альдолей, и называется альдольной конденсацией. Реакция обратима и может осуществляться только при наличии хотя бы у одного реагента атома H в a-положении к карбонильной группе. Альдольная конденсация двух различных карбонильных соединений ведет к сложным смесям. Формальдегид неспособен к димеризации, поэтому особым синтетическим приложением этого метода является соальдолизация кетонов и альдегидов с формальдегидом, например:
В концевом положении, например –CHOH–CHO или –COCH2OH, группировка взаимодействует с аммиачным раствором нитрата серебра, давая серебряное зеркало, и с ионом меди в щелочном растворе тартрата (фелингова жидкость) или цитрата (реактив Бенедикта), образуя красный осадок оксида меди(I), или медное зеркало, а также с фенилгидразином C6H5NHNH2, давая озазоны (C6H5)HNN=C(R)–CH=N–NHC6H5, которые можно гидролизовать в соответствующие a-кетоальдегиды (дикарбонильные соединения).
b-Гидроксикарбонильные соединения.
Когда ацетальдегид обрабатывают мягким основанием (например, карбонатом калия), происходит его димеризация в альдоль (b-гидроксиальдегид) CH3CHOHCH2CHO. Аналогично ацетон может димеризоваться в диацетоновой спирт (CH3)2C(OH)CH2C(O)CH3 (b-гидроксикетон). Эта реакция представляет собой метод получения b-гидроксикарбонильных соединений, или альдолей, и называется альдольной конденсацией. Реакция обратима и может осуществляться только при наличии хотя бы у одного реагента атома H в a-положении к карбонильной группе. Альдольная конденсация двух различных карбонильных соединений ведет к сложным смесям. Формальдегид неспособен к димеризации, поэтому особым синтетическим приложением этого метода является соальдолизация кетонов и альдегидов с формальдегидом, например:
 Сходная конденсация катализируется кислотами, но в этих условиях альдоль обычно дегидратируется в соответствующий a,b-ненасыщенный альдегид или кетон, например,
Сходная конденсация катализируется кислотами, но в этих условиях альдоль обычно дегидратируется в соответствующий a,b-ненасыщенный альдегид или кетон, например,
 3. Дикарбонильные соединения.
Эти вещества имеют общую формулу R–CO–(CH2)nCO–Rў и подразделяются на диальдегиды (R = Rў = H, алкандиали), кетоальдегиды (R = H, алканалоны) и дикетоны (алкандионы). Они обозначаются как a, b, g, ... дикарбонильные соединения в соответствии со значениями n = 0, 1, 2, ... .
a-Дикарбонильные соединения.
Их простейший представитель – глиоксаль CHO–CHO (этандиаль), желто-зеленое летучее вещество, получаемое окислением ацетальдегида азотной кислотой. Глиоксаль легко полимеризуется. В щелочном растворе он превращается в гликолевую кислоту HOCH2COOH. При действии аммиака и формальдегида он дает гетероциклическое соединение имидазол (см. разд. IV-4.Г).
3. Дикарбонильные соединения.
Эти вещества имеют общую формулу R–CO–(CH2)nCO–Rў и подразделяются на диальдегиды (R = Rў = H, алкандиали), кетоальдегиды (R = H, алканалоны) и дикетоны (алкандионы). Они обозначаются как a, b, g, ... дикарбонильные соединения в соответствии со значениями n = 0, 1, 2, ... .
a-Дикарбонильные соединения.
Их простейший представитель – глиоксаль CHO–CHO (этандиаль), желто-зеленое летучее вещество, получаемое окислением ацетальдегида азотной кислотой. Глиоксаль легко полимеризуется. В щелочном растворе он превращается в гликолевую кислоту HOCH2COOH. При действии аммиака и формальдегида он дает гетероциклическое соединение имидазол (см. разд. IV-4.Г).
Монозамещенные глиоксали (например, метилглиоксаль, или пропаналон-2) можно получить гидролизом соответствующих изонитрозокетонов ( R–CO–CH=NOH), которые образуются при взаимодействии алкилнитритов (R–O–N=O) с кетонами в присутствии этилата натрия. Как и глиоксаль, эти соединения склонны к полимеризации и в щелочных растворах превращаются в соответствующие a-гидроксикислоты.
Простейшим a-дикетоном является диацетил CH3COCOCH3 (бутан-2,3-дион), желтая летучая жидкость, найденная в некоторых эфирных маслах, например эфирном масле тмина. Его диоксим (диметилглиоксим) используется в аналитической химии для качественного и количественного определения иона никеля. a-Дикетоны можно получать из кетонов путем превращения последних в изонитрозокетоны:
 с последующим гидролизом изонитрозокетонов в кислой среде:
с последующим гидролизом изонитрозокетонов в кислой среде:
 a-Дикетоны можно также получить конденсацией Клайзена из эфиров щавелевой кислоты:
a-Дикетоны можно также получить конденсацией Клайзена из эфиров щавелевой кислоты:
 a-Дикетоны являются полезными промежуточными соединениями в синтезе некоторых гетероциклических веществ (см. ниже). Так, при действии аммиака и альдегида из них образуются производные имидазола. Обработка a-диаминами дает пиперазины.
b-Дикетоны.
Самый общий путь получения этих соединений – клайзеновская конденсация сложных эфиров с кетонами:
a-Дикетоны являются полезными промежуточными соединениями в синтезе некоторых гетероциклических веществ (см. ниже). Так, при действии аммиака и альдегида из них образуются производные имидазола. Обработка a-диаминами дает пиперазины.
b-Дикетоны.
Самый общий путь получения этих соединений – клайзеновская конденсация сложных эфиров с кетонами:
 b-Дикетоны представляют собой слабые кислоты, поскольку атомы водорода метиленовой группы сильно активируются соседними карбонильными группами. Натриевые соли дикетонов при обработке алкилгалогенидами дают алкилзамещенные b-дикетоны:
b-Дикетоны представляют собой слабые кислоты, поскольку атомы водорода метиленовой группы сильно активируются соседними карбонильными группами. Натриевые соли дикетонов при обработке алкилгалогенидами дают алкилзамещенные b-дикетоны:
 Простейший член ряда – ацетилацетон (пентан-2,4-дион) легко образует комплексные хелатные соли со многими металлами, например,
Простейший член ряда – ацетилацетон (пентан-2,4-дион) легко образует комплексные хелатные соли со многими металлами, например,
 Такие соли обычно интенсивно окрашены, растворимы в хлороформе и других неполярных растворителях, некоторые из них (соли бериллия, алюминия) летучи. b-Дикетоны чувствительны к действию водных щелочей, которые расщепляют их на кетоны и кислоты. Их часто используют как промежуточные соединения в синтезе пиразолов.
g-Дикетоны
можно получить действием алкилцинкгалогенидов на сукцинилхлорид:
Такие соли обычно интенсивно окрашены, растворимы в хлороформе и других неполярных растворителях, некоторые из них (соли бериллия, алюминия) летучи. b-Дикетоны чувствительны к действию водных щелочей, которые расщепляют их на кетоны и кислоты. Их часто используют как промежуточные соединения в синтезе пиразолов.
g-Дикетоны
можно получить действием алкилцинкгалогенидов на сукцинилхлорид:
 или из b-кетоэфиров:
или из b-кетоэфиров:
 Они полезны для синтеза некоторых гетероциклических соединений (см. разд. IV).
4. Кислоты, содержащие еще одну функциональную (некарбоксильную) группу.
Галогенокислоты.
Из галогенокислот только a-галогенокислоты R–CHX–COOH можно получить прямым галогенированием, обычно обработкой кислот или соответствующих хлорангидридов (RCH2COCl) хлором или бромом в присутствии красного фосфора. Образующиеся a-галогенацилгалогениды легко гидролизуются до свободных кислот. b-Бромкислоты обычно готовят присоединением бромоводорода к a,b-ненасыщенной кислоте:
Они полезны для синтеза некоторых гетероциклических соединений (см. разд. IV).
4. Кислоты, содержащие еще одну функциональную (некарбоксильную) группу.
Галогенокислоты.
Из галогенокислот только a-галогенокислоты R–CHX–COOH можно получить прямым галогенированием, обычно обработкой кислот или соответствующих хлорангидридов (RCH2COCl) хлором или бромом в присутствии красного фосфора. Образующиеся a-галогенацилгалогениды легко гидролизуются до свободных кислот. b-Бромкислоты обычно готовят присоединением бромоводорода к a,b-ненасыщенной кислоте:
 Галогенокислоты с атомом галогена, более удаленным от карбоксильной группы, получают действием бромо- или хлороводорода на соответствующие гидроксикислоты. Натриевые соли этих кислот в слабощелочных растворах легко гидролизуются до натриевых солей соответствующих гидроксикислот. Обработка галогенокислот аммиаком дает аминокислоты.
Гидроксикислоты (оксокислоты).
a-Гидроксикислоты можно получить гидролизом галогенокислот или из альдегидов (кетонов), используя синтез Килиани:
Галогенокислоты с атомом галогена, более удаленным от карбоксильной группы, получают действием бромо- или хлороводорода на соответствующие гидроксикислоты. Натриевые соли этих кислот в слабощелочных растворах легко гидролизуются до натриевых солей соответствующих гидроксикислот. Обработка галогенокислот аммиаком дает аминокислоты.
Гидроксикислоты (оксокислоты).
a-Гидроксикислоты можно получить гидролизом галогенокислот или из альдегидов (кетонов), используя синтез Килиани:
 Гликолевая кислота CH2OH–COOH и молочная кислота CH3–CHOH–COOH встречаются в природе. Последняя образуется в результате молочнокислого брожения (при скисании молока) и обнаружена в тканях животных, растений и в микроорганизмах как конечный продукт окисления сахаров.
b-Гидрооксикислоты можно получить кислотно-катализируемым присоединением воды к a,b-ненасыщенным кислотам. Их эфиры можно приготовить из альдегидов и кетонов по реакции Реформатского:
Гликолевая кислота CH2OH–COOH и молочная кислота CH3–CHOH–COOH встречаются в природе. Последняя образуется в результате молочнокислого брожения (при скисании молока) и обнаружена в тканях животных, растений и в микроорганизмах как конечный продукт окисления сахаров.
b-Гидрооксикислоты можно получить кислотно-катализируемым присоединением воды к a,b-ненасыщенным кислотам. Их эфиры можно приготовить из альдегидов и кетонов по реакции Реформатского:
 b-Гидроксимасляную кислоту иногда находят в виде левовращающей оптически активной формы в моче, особенно у больных сахарным диабетом. b-Гидроксикислоты довольно неустойчивы, особенно к действию сильных неорганических кислот; они легко теряют воду, образуя a,b-ненасыщенные кислоты.
g-Гидроксикислоты редко встречаются в свободном состоянии; они обычно самопроизвольно переходят в устойчивые g-лактоны, которые можно получить также кислотно-катализируемой изомеризацией b,g-ненасыщенных кислот:
b-Гидроксимасляную кислоту иногда находят в виде левовращающей оптически активной формы в моче, особенно у больных сахарным диабетом. b-Гидроксикислоты довольно неустойчивы, особенно к действию сильных неорганических кислот; они легко теряют воду, образуя a,b-ненасыщенные кислоты.
g-Гидроксикислоты редко встречаются в свободном состоянии; они обычно самопроизвольно переходят в устойчивые g-лактоны, которые можно получить также кислотно-катализируемой изомеризацией b,g-ненасыщенных кислот:
 Эфиры g-гидроксикислот можно приготовить восстановлением соответствующих эфиров кетокислот.
Аминокислоты.
Наиболее важными членами этой группы являются a-аминокислоты:
Эфиры g-гидроксикислот можно приготовить восстановлением соответствующих эфиров кетокислот.
Аминокислоты.
Наиболее важными членами этой группы являются a-аминокислоты:
 Многие из них можно получить гидролизом белков, в которых они, как оказалось, соединены в длинные пептидные цепи типа
Многие из них можно получить гидролизом белков, в которых они, как оказалось, соединены в длинные пептидные цепи типа
 Все природные аминокислоты (кроме глицина H2NCH2COOH) оптически активны, и им приписана конфигурация
Все природные аминокислоты (кроме глицина H2NCH2COOH) оптически активны, и им приписана конфигурация
 обозначаемая как L (levo). Группа R в природных аминокислотах может быть алифатической (например, в аланине R = –CH3, в серине R = –CH2OH, в цистеине R = –CH2SH, в лейцине R = –CH2CH(CH3)2), ароматической (как в фенилаланине R = –CH2C6H5 и тирозине R = –CH2C6H4OH) или гетероциклической, как в гистидине
обозначаемая как L (levo). Группа R в природных аминокислотах может быть алифатической (например, в аланине R = –CH3, в серине R = –CH2OH, в цистеине R = –CH2SH, в лейцине R = –CH2CH(CH3)2), ароматической (как в фенилаланине R = –CH2C6H5 и тирозине R = –CH2C6H4OH) или гетероциклической, как в гистидине
 Аминокислоты – кристаллические вещества, сильно различающиеся по растворимости в воде. Хотя все они обладают L-конфигурацией, некоторые из них являются правовращающими (+), а некоторые – левовращающими (–). Из гидролизатов белков выделено 20 аминокислот. Качественный и количественный аминокислотный состав сильно различается для разных белков.
Аминокислоты можно получить действием аммиака на a-галогенокислоты или при помощи синтеза Штреккера (модификации синтеза Килиани), при котором альдегид обрабатывают синильной кислотой в присутствии хлорида аммония. Образующиеся a-аминонитрилы RCHNH2CN можно затем гидролизовать до желаемых кислот. Такие синтетические аминокислоты оптически неактивны и состоят из равных количеств право- и левовращающих форм. Разделения на оптические антиподы часто можно достичь фракционной кристаллизацией их солей с такими оптически активными основаниями, как бруцин.
Аминокислоты – кристаллические вещества, сильно различающиеся по растворимости в воде. Хотя все они обладают L-конфигурацией, некоторые из них являются правовращающими (+), а некоторые – левовращающими (–). Из гидролизатов белков выделено 20 аминокислот. Качественный и количественный аминокислотный состав сильно различается для разных белков.
Аминокислоты можно получить действием аммиака на a-галогенокислоты или при помощи синтеза Штреккера (модификации синтеза Килиани), при котором альдегид обрабатывают синильной кислотой в присутствии хлорида аммония. Образующиеся a-аминонитрилы RCHNH2CN можно затем гидролизовать до желаемых кислот. Такие синтетические аминокислоты оптически неактивны и состоят из равных количеств право- и левовращающих форм. Разделения на оптические антиподы часто можно достичь фракционной кристаллизацией их солей с такими оптически активными основаниями, как бруцин.
Аминокислоты амфотерны; в реакциях они проявляют себя то как слабые основания  , то как слабые кислоты , то как слабые кислоты  . Восстановление эфиров аминокислот дает a-аминоспирты. При высоких температурах многие аминокислоты теряют диоксид углерода, образуя амины. Действие азотистой кислоты ведет к образованию соответствующих a-гидроксикислот, а действие нитрозилгалогенидов – к образованию a-галогенокислот. . Восстановление эфиров аминокислот дает a-аминоспирты. При высоких температурах многие аминокислоты теряют диоксид углерода, образуя амины. Действие азотистой кислоты ведет к образованию соответствующих a-гидроксикислот, а действие нитрозилгалогенидов – к образованию a-галогенокислот.
Многие из этих реакций осуществляются в более мягких условиях под действием ферментных систем живых организмов. Так, Bacillus proteus, B. coli и B. subtilis декарбоксилируют многие природные аминокислоты до аминов. Такие амины (например, путресцин H2N–(CH2)4–NH2) являются характерными продуктами разложения белков. Эти бактерии способны также заменять аминогруппу на гидроксил в мягких щелочных условиях.
5. Кетокислоты (оксокислоты, альдегидокислоты).
a-Кетокислоты.
Простейшая альдегидокислота – глиоксалевая кислота HC(=O)–COOH – может быть получена гидролизом дихлоруксусной кислоты или электролитическим восстановлением щавелевой кислоты. Она встречается в природе в незрелых плодах. Простейшая a-кетокислота – пировиноградная кислота CH3COCOOH – является важным промежуточным веществом в окислительном и ферментативном расщеплении сахаров (в гликолизе) живыми организмами. Ее можно получить, используя общие реакции, ведущие к образованию a-кетокислот:
 a-Кетокислоты легко окисляются:
a-Кетокислоты легко окисляются:
 Они также очень чувствительны к действию концентрированной серной кислоты, вызывающей реакцию
Они также очень чувствительны к действию концентрированной серной кислоты, вызывающей реакцию
 b-Кетокислоты.
Свободные b-кетокислоты при нагревании легко выделяют углекислый газ, образуя кетоны:
b-Кетокислоты.
Свободные b-кетокислоты при нагревании легко выделяют углекислый газ, образуя кетоны:
 Однако их эфиры вполне устойчивы. Их можно получить из сложных эфиров конденсацией Клайзена:
Однако их эфиры вполне устойчивы. Их можно получить из сложных эфиров конденсацией Клайзена:
 Их простейший представитель – ацетоуксусный эфир CH3COCH2COOC2H5 – имеет большое значение для органического синтеза. Его натриевая соль CH3–CO–CHNa–COOC2H5, получаемая действием этилата натрия, легко реагирует с алкилгалогенидами, давая алкилацетоуксусные эфиры, CH3COCHRCOOC2H5. Их в свою очередь можно проалкилировать до диалкилпроизводных CH3COCRRўCOOC2H5. Расщепление таких эфиров до замещенных уксусных кислот можно осуществить действием сильных щелочей:
Их простейший представитель – ацетоуксусный эфир CH3COCH2COOC2H5 – имеет большое значение для органического синтеза. Его натриевая соль CH3–CO–CHNa–COOC2H5, получаемая действием этилата натрия, легко реагирует с алкилгалогенидами, давая алкилацетоуксусные эфиры, CH3COCHRCOOC2H5. Их в свою очередь можно проалкилировать до диалкилпроизводных CH3COCRRўCOOC2H5. Расщепление таких эфиров до замещенных уксусных кислот можно осуществить действием сильных щелочей:
 Расщепление до замещенных ацетонов достигается обработкой разбавленными кислотами или щелочами:
Расщепление до замещенных ацетонов достигается обработкой разбавленными кислотами или щелочами:
 С реагентами, специфическими для карбонильной группы (см. разд. IV-1.А.4), b-кетоэфиры часто дают гетероциклы (см. разд. IV-4). Так, действие фенилгидразина C6H5NHNH2 ведет к пиразолонам, а гидроксиламина – к изоксазолонам.
6. Дикарбоновые кислоты
принадлежат к гомологическому ряду HOOC(CH2)nCOOH (n = 0, 1, 2, 3, ...), бльшая часть членов которого встречается в природе.
Щавелевая кислота
HOOC–COOH широко распространена в растениях в форме ее солей (натрия, калия, кальция и магния). Натриевую соль получают в промышленности действием углекислого газа на натрий при 360° С. Это довольно сильная кислота (K1 = 0,04). Серная кислота превращает ее в углекислый газ, монооксид углерода и воду. Действием перманганата калия в горячем кислом растворе она количественно превращается в CO2. С ионами переходных элементов оксалат-ион образует многочисленные устойчивые комплексные ионы, например [Fe(C2O4)3]3–; [Co(C2O4)3]3–; [Cr(C2O4)3]3–. Последние два иона получены в оптически активной форме, что можно объяснить, только если связи металл-кислород являются по сути ковалентными и направлены к вершинам правильного октаэдра.
Малоновая кислота
HOOC–CH2–COOH найдена в свекольном соке. Ее можно получить из хлоруксусной кислоты, первоначально превратив последнюю в цианоуксусную кислоту NC–CH2–COOH действием NaCN с последующим гидролизом. Свободная малоновая кислота и ее C-алкилпроизводные при нагревании выше 140° С теряют CO2, давая монокарбоновые кислоты. Ее эфиры, как и ацетоуксусный эфир, легко алкилируются путем превращения в натриевую соль с последующей обработкой алкилгалогенидом:
С реагентами, специфическими для карбонильной группы (см. разд. IV-1.А.4), b-кетоэфиры часто дают гетероциклы (см. разд. IV-4). Так, действие фенилгидразина C6H5NHNH2 ведет к пиразолонам, а гидроксиламина – к изоксазолонам.
6. Дикарбоновые кислоты
принадлежат к гомологическому ряду HOOC(CH2)nCOOH (n = 0, 1, 2, 3, ...), бльшая часть членов которого встречается в природе.
Щавелевая кислота
HOOC–COOH широко распространена в растениях в форме ее солей (натрия, калия, кальция и магния). Натриевую соль получают в промышленности действием углекислого газа на натрий при 360° С. Это довольно сильная кислота (K1 = 0,04). Серная кислота превращает ее в углекислый газ, монооксид углерода и воду. Действием перманганата калия в горячем кислом растворе она количественно превращается в CO2. С ионами переходных элементов оксалат-ион образует многочисленные устойчивые комплексные ионы, например [Fe(C2O4)3]3–; [Co(C2O4)3]3–; [Cr(C2O4)3]3–. Последние два иона получены в оптически активной форме, что можно объяснить, только если связи металл-кислород являются по сути ковалентными и направлены к вершинам правильного октаэдра.
Малоновая кислота
HOOC–CH2–COOH найдена в свекольном соке. Ее можно получить из хлоруксусной кислоты, первоначально превратив последнюю в цианоуксусную кислоту NC–CH2–COOH действием NaCN с последующим гидролизом. Свободная малоновая кислота и ее C-алкилпроизводные при нагревании выше 140° С теряют CO2, давая монокарбоновые кислоты. Ее эфиры, как и ацетоуксусный эфир, легко алкилируются путем превращения в натриевую соль с последующей обработкой алкилгалогенидом:
 Малоновая кислота, таким образом, очень полезна для синтеза кислот. Действуя на натриевые соли ее эфиров дигалогенидами (или галогеном), можно получить высшие дикарбоновые кислоты:
Малоновая кислота, таким образом, очень полезна для синтеза кислот. Действуя на натриевые соли ее эфиров дигалогенидами (или галогеном), можно получить высшие дикарбоновые кислоты:
 Активные атомы водорода метиленовой группы сходным образом участвуют в реакциях с карбонильными соединениями. Так, с альдегидами в присутствии пиридина как катализатора получаются a,b-ненасыщенные кислоты (реакция Кнёвенагеля):
Активные атомы водорода метиленовой группы сходным образом участвуют в реакциях с карбонильными соединениями. Так, с альдегидами в присутствии пиридина как катализатора получаются a,b-ненасыщенные кислоты (реакция Кнёвенагеля):
 Если вместо пиридина используется триэтаноламин, продуктами реакции являются b,g-ненасыщенные кислоты (модификация Линстеда).
Янтарная кислота
HOOC–CH2–CH2–COOH широко распространена в природе (свекольный сок, янтарь, виноград и т.д.) и образуется во многих процессах брожения. Ее получают главным образом сбраживанием тартрата аммония и малата кальция (см. ниже обсуждение яблочной кислоты под заголовками «Ненасыщенные дикарбоновые кислоты» и «Гидрокси- и кетодикарбоновые кислоты»). Ее можно получить и синтетически гидролизом ее динитрила NCCH2CH2CN, который в свою очередь получают действием цианида калия на дибромэтан. Ее ангидрид
Если вместо пиридина используется триэтаноламин, продуктами реакции являются b,g-ненасыщенные кислоты (модификация Линстеда).
Янтарная кислота
HOOC–CH2–CH2–COOH широко распространена в природе (свекольный сок, янтарь, виноград и т.д.) и образуется во многих процессах брожения. Ее получают главным образом сбраживанием тартрата аммония и малата кальция (см. ниже обсуждение яблочной кислоты под заголовками «Ненасыщенные дикарбоновые кислоты» и «Гидрокси- и кетодикарбоновые кислоты»). Ее можно получить и синтетически гидролизом ее динитрила NCCH2CH2CN, который в свою очередь получают действием цианида калия на дибромэтан. Ее ангидрид  , который образуется при нагревании кислоты с ацетилхлоридом, часто используется как этерифицирующий агент, особенно если желательно отделить спирт от других веществ. Полученные таким образом полуэфиры янтарной кислоты ROOCCH2CH2COOH благодаря их кислотным свойствам могут быть легко отделены от нейтральных примесей. Регенерация спирта осуществляется омылением. , который образуется при нагревании кислоты с ацетилхлоридом, часто используется как этерифицирующий агент, особенно если желательно отделить спирт от других веществ. Полученные таким образом полуэфиры янтарной кислоты ROOCCH2CH2COOH благодаря их кислотным свойствам могут быть легко отделены от нейтральных примесей. Регенерация спирта осуществляется омылением.
Глутаровая кислота
HOOC(CH2)3COOH содержится в свекольном соке и смывах с неочищенной шерсти. Ее можно получить из малонового эфира общими реакциями, приведенными выше.
Адипиновая кислота
HOOC(CH2)4COOH получается окислением касторового масла. Ее можно легко приготовить окислением циклогексанола (см. разд. IV-2.А.2) азотной кислотой в присутствии ванадиевого катализатора. При пиролизе этой кислоты и ее С-алкильных производных, особенно в присутствии оксидов кальция, бария и тория, образуются циклопентаноны (см. разд. IV-2.А.1). Она используется для получения найлона.
Ненасыщенные дикарбоновые кислоты.
Малеиновая и фумаровая кислоты представляют собой соответственно цис- и транс-формы дегидроянтарной кислоты HOOCCH=CHCOOH и получаются при нагревании яблочной кислоты (гидроксиянтарная кислота, см. ниже).
 Фумаровая кислота является главным продуктом при более низких температурах, малеиновая – при более высоких. Из этих двух кислот только малеиновая образует ангидрид, что свидетельствует о ее цис-конфигурации, тогда как в фумаровой кислоте карбоксильные группы слишком удалены друг от друга. Фумаровая кислота встречается в природе в дымянке (Fumaria officinalis) и других растениях.
При термическом разложении лимонной кислоты (см. разд. IV-1.Б.7) образуются три изомерные кислоты C5H6O4:
Фумаровая кислота является главным продуктом при более низких температурах, малеиновая – при более высоких. Из этих двух кислот только малеиновая образует ангидрид, что свидетельствует о ее цис-конфигурации, тогда как в фумаровой кислоте карбоксильные группы слишком удалены друг от друга. Фумаровая кислота встречается в природе в дымянке (Fumaria officinalis) и других растениях.
При термическом разложении лимонной кислоты (см. разд. IV-1.Б.7) образуются три изомерные кислоты C5H6O4:
 Гидрокси- (
Гидрокси- ( окси-) и кетодикарбоновые кислоты.
Тартроновая (оксималоновая) кислота HOOC–CHOH–COOH получается в виде эфира при действии оксида серебра на броммалоновый эфир. Первым продуктом ее окисления является мезоксалевая кислота HOOC–CO–COOH.
Яблочная (оксиянтарная) кислота HOOC–CHOH–CH2–COOH встречается в природе в виде оптически активной левовращающей формы во многих плодах. Мягкое окисление превращает ее в щавелевоуксусную кислоту HOOC–CO–CH2–COOH, существующую в виде цис,транс-изомерных форм.
Винная кислота HOOC–CHOH–CHOH–COOH известна в четырех формах: две из них являются оптически активными зеркальными (+)- и (–)-изомерами (т. пл. 170° С); рацемическая (±)-винная (виноградная) кислота (т. пл. 204° С) представляет собой эквимолярный молекулярный комплекс (+)- и (–)-форм; мезовинная кислота (т. пл. 140° С) обладает плоскостью симметрии и потому оптически неактивна. (+)-Винная кислота получается из вина в виде ее кислой калиевой соли. При обработке щелочью она превращается в смесь (+)-, (–)- и мезо-форм.
7. Трикарбоновые кислоты.
Трикарбаллиловая кислота
HOOC–CH2–CH(COOH)–CH2–COOH получается гидролизом ее тринитрила NC–CH2–CH(CN)–CH2–CN, образующегося при действии цианида калия на 1,2,3-трибромпропан. Она также является продуктом гидрирования аконитовой кислоты, возникающей при дегидратации лимонной кислоты.
Лимонная кислота
HOOC–CH2–C(OH)(COOH)–CH2–COOH найдена во многих фруктовых соках. Ее получают в промышленных масштабах действием микроорганизмов (цитромицетов) на такие углеводы, как мальтоза. Она находит многочисленные применения в приготовлении искусственных фруктовых напитков. Ее дегидратация дает аконитовую кислоту HOOC–CH=C(COOH)–CH2–COOH.
IV-2. АЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Алициклические соединения определяют как карбоциклические (в цикле присутствуют только атомы углерода) соединения, которые в своих реакциях обнаруживают преимущественно алифатический характер в противоположность поведению ароматических соединений. Их удобно подразделить по числу колец в молекуле на моноциклические (одноядерные) и полициклические (многоядерные – бициклические, трициклические и т.д.) кольцевые системы. Многие насыщенные алициклические углеводороды найдены в нефти. Ненасыщенные алициклические углеводороды, возникающие, по крайней мере формально, при полимеризации изопрена CH2=C(CH3)CH=CH2 и известные под названием терпенов, в изобилии встречаются во многих эфирных маслах. Алициклические системы часто встречаются в природных соединениях, иных, чем терпены, например в каротиноидах и стероидах.
А. МОНОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
1. Синтез.
Следующие реакции иллюстрируют общие методы синтеза. Обычно пяти- и шестичленные кольца образуются легче всего, так как углерод может сохранить в них нормальные валентные углы.
1) Из дигалогенидов:
 2) Из малонового или ацетоуксусного эфира:
2) Из малонового или ацетоуксусного эфира:

 3) Конденсация эфиров двухосновных кислот (реакция Дикмана):
3) Конденсация эфиров двухосновных кислот (реакция Дикмана):
 В развитие метода получения кетонов с большими циклами разработан метод внутримолекулярной ацилоиновой конденсации путем кипячения эфиров дикарбоновых кислот с металлическим натрием; образующиеся оксикетоны могут быть легко превращены в циклические кетоны.
4) Внутримолекулярная альдольная конденсация некоторых дикетонов:
В развитие метода получения кетонов с большими циклами разработан метод внутримолекулярной ацилоиновой конденсации путем кипячения эфиров дикарбоновых кислот с металлическим натрием; образующиеся оксикетоны могут быть легко превращены в циклические кетоны.
4) Внутримолекулярная альдольная конденсация некоторых дикетонов:
 5) Сухая перегонка кальциевых, бариевых или ториевых солей адипиновой кислоты (n = 4) и ее высших гомологов:
5) Сухая перегонка кальциевых, бариевых или ториевых солей адипиновой кислоты (n = 4) и ее высших гомологов:
 6) (2 + 2)-циклоприсоединением получают 4-членные циклы:
6) (2 + 2)-циклоприсоединением получают 4-членные циклы:
 7) Диеновый синтез используется для получения 6-членных циклов:
7) Диеновый синтез используется для получения 6-членных циклов:
 Существует еще много других методов синтеза алициклических соединений, среди них гидрирование производных бензола, внутримолекулярная конденсация g- и d-дикетонов, полимеризация ацетилена в особых условиях с образованием 8- и 10-членных циклов, прямая циклизация насыщенных углеводородов и т.д. Кроме того, возможны превращения циклов друг в друга, при этом может происходить расширение или сужение цикла.
2. Расширение и сужение цикла.
Методы расширения и сужения цикла главным образом основаны на способности циклических систем претерпевать молекулярные перегруппировки. В связи с этим особенно широко используется перегруппировка Вагнера – Мейервейна и родственные перегруппировки (RX + Y ® RўY + X), сопровождающие многие реакции замещения в алифатическом ряду, в которых в качестве промежуточных частиц возникают неустойчивые ионы R+.
Ниже следуют примеры:
Существует еще много других методов синтеза алициклических соединений, среди них гидрирование производных бензола, внутримолекулярная конденсация g- и d-дикетонов, полимеризация ацетилена в особых условиях с образованием 8- и 10-членных циклов, прямая циклизация насыщенных углеводородов и т.д. Кроме того, возможны превращения циклов друг в друга, при этом может происходить расширение или сужение цикла.
2. Расширение и сужение цикла.
Методы расширения и сужения цикла главным образом основаны на способности циклических систем претерпевать молекулярные перегруппировки. В связи с этим особенно широко используется перегруппировка Вагнера – Мейервейна и родственные перегруппировки (RX + Y ® RўY + X), сопровождающие многие реакции замещения в алифатическом ряду, в которых в качестве промежуточных частиц возникают неустойчивые ионы R+.
Ниже следуют примеры:
1) Диазотирование циклических аминов (реакция Демьянова):
 2) Действие щелочи на 2-галогеноциклоалканоны (перегруппировка Фаворского):
2) Действие щелочи на 2-галогеноциклоалканоны (перегруппировка Фаворского):
 3) Пинаколиновая перегруппировка циклических гликолей:
3) Пинаколиновая перегруппировка циклических гликолей:
 4) Многие циклопарафины можно мягко и обратимо изомеризовать над хлоридом алюминия, например:
4) Многие циклопарафины можно мягко и обратимо изомеризовать над хлоридом алюминия, например:
 3. Стереоизомерия
(см. разд. II.Б, «Оптическая изомерия») алициклических соединений включает как геометрическую (цис-транс), так и оптическую изомерию. Явление геометрической изомерии возникает всякий раз, когда два или более заместителей присоединены к кольцу, поскольку атомы углерода кольца не могут свободно вращаться один относительно другого. Когда изомер не может быть совмещен со своим зеркальным изображением, имеет место оптическая изомерия, т.е. его можно разделить на право- и левовращающую формы. Если же изомер обладает плоскостью симметрии, он не может быть разделен на энантиомеры.
3. Стереоизомерия
(см. разд. II.Б, «Оптическая изомерия») алициклических соединений включает как геометрическую (цис-транс), так и оптическую изомерию. Явление геометрической изомерии возникает всякий раз, когда два или более заместителей присоединены к кольцу, поскольку атомы углерода кольца не могут свободно вращаться один относительно другого. Когда изомер не может быть совмещен со своим зеркальным изображением, имеет место оптическая изомерия, т.е. его можно разделить на право- и левовращающую формы. Если же изомер обладает плоскостью симметрии, он не может быть разделен на энантиомеры.
Когда алициклическое кольцо несет различные заместители на каждом из n углеродных атомов, возможно 2n – 1 геометрических изомеров. Любой из этих изомеров, если он асимметричен, может быть разделен на право- и левовращающую формы. Когда два или более заместителей одинаковы, вследствие симметрии достаточно часто число изомеров оказывается меньше теоретического максимума. Так, инозит (CHOH)6 (см. разд. IV-2.А.6) имеет только 8 изомеров, и лишь один из них можно разделить на правую и левую формы. Проблема стереоизомерии алициклических соединений играет решающую роль при определении структуры физиологически активных природных соединений и в их синтезе. Часто только один изомер из сотен теоретически возможных имеет заметную физиологическую активность.
4. Реакции.
Между реакциями алифатических и алициклических соединений наблюдается существенное сходство. Те реакции, которые требуют подхода внешнего реагента (например, реакции присоединения по карбонилу, реакции замещения), часто протекают быстрее с алициклическими, чем с алифатическими соединениями, содержащими то же число углеродных атомов. Эта ситуация возникает в результате жесткости кольца, ограничивающей стерические препятствия, обусловленные неупорядоченным движением заместителей, присоединенных к атакуемому атому углерода. В то же время, внутримолекулярные реакции могут ускоряться или затрудняться благоприятным или неблагоприятным расположением реагирующих групп. Так, цис-дикарбоновые кислоты, как правило, легко образуют ангидриды, тогда как у транс-изомеров это превращение обычно затруднено или просто невозможно. Цис-гидроксикислоты легко образуют лактоны, а транс-изомеры – нет. Отщепление элементов галогеноводорода под действием сильных оснований с образованием олефина требует транс-расположения отщепляющихся H и X. Соответственно водород в транс-положении к соседнему галогену достаточно лабилен, а в цис-положении необычайно стабилен. Из-за сходства алициклических и алифатических соединений детальное обсуждение отдельных алициклов будет опущено.
Особую группу реакций составляют реакции разрыва колец (раскрытия цикла):
1) гидрогенолиз:
 2) окисление циклоолефинов:
2) окисление циклоолефинов:
 3) окислительное расщепление циклических гликолей под действием HIO4 (см. разд. IV-1.Б.1):
3) окислительное расщепление циклических гликолей под действием HIO4 (см. разд. IV-1.Б.1):
 4) окисление циклических кетонов азотной кислотой (см. разд. IV-1.Б.6, «Адипиновая кислота»);
4) окисление циклических кетонов азотной кислотой (см. разд. IV-1.Б.6, «Адипиновая кислота»);
5) бекмановская перегруппировка циклических кетоксимов:
 Ниже рассмотрены примеры наиболее важных природных соединений циклопентанового и циклогексанового рядов.
5. Природные производные циклопентана.
Некоторые природные душистые вещества, например жасмон из масла жасмина, содержат циклопентановую структуру
Ниже рассмотрены примеры наиболее важных природных соединений циклопентанового и циклогексанового рядов.
5. Природные производные циклопентана.
Некоторые природные душистые вещества, например жасмон из масла жасмина, содержат циклопентановую структуру
 Из кислот ряда циклопентана хаульмугровая кислота
Из кислот ряда циклопентана хаульмугровая кислота
 из масла хаульмугровой пальмы применяется для лечения проказы. Ауксины а и б, факторы роста растений, являются ненасыщенными гидроксикислотами, производными циклопентана:
из масла хаульмугровой пальмы применяется для лечения проказы. Ауксины а и б, факторы роста растений, являются ненасыщенными гидроксикислотами, производными циклопентана:
 6. Природные производные циклогексана.
Терпены,
являющиеся производными циклогексана, достаточно часто встречаются в природе, особенно в эфирных маслах. Наиболее обычны моноциклические терпены со скелетом ментана:
6. Природные производные циклогексана.
Терпены,
являющиеся производными циклогексана, достаточно часто встречаются в природе, особенно в эфирных маслах. Наиболее обычны моноциклические терпены со скелетом ментана:
 Ментадиены C10H16 можно рассматривать как продукты конденсации двух молекул изопрена CH2=C(CH3)–CH=CH2. Для них возможны многочисленные изомеры, существование которых обусловлено различным расположением двойных связей (D) и оптической изомерией. Типичным примером является лимонен (D1,8), который встречается в виде D-формы в маслах апельсина и тмина, в виде L-формы в сосновом и еловом масле и в рацемической форме (дипентен) в скипидаре. Из ментенов C10H18 наиболее важен D3-ментен (из масла тмина), который может быть получен дегидратацией ментола (см. ниже). Многие спирты, например ментол (ментанол-3 из масла перечной мяты) и a-терпинеол (D1-ментенол-8 из масел кардамона и майорана), являются производными этих углеводородов. Двухатомные спирты этого ряда известны под названием терпины. Из них наибольший интерес представляет терпингидрат (ментандиол-1,8), который образуется во многих эфирных маслах при стоянии.
Среди кетонов этого ряда заслуживают упоминания изомеры ментона-3 и пулегона (D4(8)-ментенона-3), которые содержатся в мятном масле, и карвон (D1,8-ментадиенон-6), найденный в маслах тмина и укропа.
Ментадиены C10H16 можно рассматривать как продукты конденсации двух молекул изопрена CH2=C(CH3)–CH=CH2. Для них возможны многочисленные изомеры, существование которых обусловлено различным расположением двойных связей (D) и оптической изомерией. Типичным примером является лимонен (D1,8), который встречается в виде D-формы в маслах апельсина и тмина, в виде L-формы в сосновом и еловом масле и в рацемической форме (дипентен) в скипидаре. Из ментенов C10H18 наиболее важен D3-ментен (из масла тмина), который может быть получен дегидратацией ментола (см. ниже). Многие спирты, например ментол (ментанол-3 из масла перечной мяты) и a-терпинеол (D1-ментенол-8 из масел кардамона и майорана), являются производными этих углеводородов. Двухатомные спирты этого ряда известны под названием терпины. Из них наибольший интерес представляет терпингидрат (ментандиол-1,8), который образуется во многих эфирных маслах при стоянии.
Среди кетонов этого ряда заслуживают упоминания изомеры ментона-3 и пулегона (D4(8)-ментенона-3), которые содержатся в мятном масле, и карвон (D1,8-ментадиенон-6), найденный в маслах тмина и укропа.
Терпены представляют собой летучие приятно пахнущие жидкости; их применяют как отдушки и в производстве духов. Для парфюмерной промышленности большое значение имеют два родственных терпенам кетона, а именно a- и b-иононы. Они обладают запахом фиалок. Их смесь получают синтетически альдольной конденсацией ацетона с цитралем (CH3)2C=CH–(CH2)2–C(CH3)=CH–CH=O.
 Полигидроксипроизводные циклогексана.
Некоторые природные полигидроксициклогексаны в химическом отношении занимают среднее положение между сахарами и таннинами (см. разд. IV-3.А.2, «Ароматические кислоты»). Выше уже упоминался гексаатомный спирт инозит. Одна из неактивных форм, называемая мезо-инозитом, широко распространена в растительных и животных тканях. Метиловые эфиры D- и L-форм находятся соответственно в Pinus lambertiana и в коре квебрахо. Кверцит (циклогексанпентол) встречается в правовращающей форме в желудях. Хининовые алкалоиды находятся в хинной корке в соединении с хинной (1,3,4,5-тетрагидроксициклогексанкарбоновой) кислотой. Эта кислота при дегидратации дает шикимовую кислоту (3,4,5-D1-тригидроксициклогексенкарбоновую кислоту), которая найдена в плодах японского звездчатого аниса (бадьяна).
7. Макроциклические системы.
Алициклические соединения с бльшими, чем циклогексан, кольцами встречаются редко и имеют относительно небольшое значение. Исключениями являются очень большие кольца пахучих начал циветты и мускуса, например:
Полигидроксипроизводные циклогексана.
Некоторые природные полигидроксициклогексаны в химическом отношении занимают среднее положение между сахарами и таннинами (см. разд. IV-3.А.2, «Ароматические кислоты»). Выше уже упоминался гексаатомный спирт инозит. Одна из неактивных форм, называемая мезо-инозитом, широко распространена в растительных и животных тканях. Метиловые эфиры D- и L-форм находятся соответственно в Pinus lambertiana и в коре квебрахо. Кверцит (циклогексанпентол) встречается в правовращающей форме в желудях. Хининовые алкалоиды находятся в хинной корке в соединении с хинной (1,3,4,5-тетрагидроксициклогексанкарбоновой) кислотой. Эта кислота при дегидратации дает шикимовую кислоту (3,4,5-D1-тригидроксициклогексенкарбоновую кислоту), которая найдена в плодах японского звездчатого аниса (бадьяна).
7. Макроциклические системы.
Алициклические соединения с бльшими, чем циклогексан, кольцами встречаются редко и имеют относительно небольшое значение. Исключениями являются очень большие кольца пахучих начал циветты и мускуса, например:
 Б. БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
1. Бициклические терпены.
Наиболее часто встречаются бициклические терпены, которые можно рассматривать как производные следующих четырех углеводородов:
Б. БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
1. Бициклические терпены.
Наиболее часто встречаются бициклические терпены, которые можно рассматривать как производные следующих четырех углеводородов:
 Можно заметить, что туйановая система включает метиленовый мостик. В пинане, каране и камфане имеется изопропилиденовый мостик
Можно заметить, что туйановая система включает метиленовый мостик. В пинане, каране и камфане имеется изопропилиденовый мостик  . Циклопропановые кольца в производных карана и туйана и циклобутановое кольцо в производных пинана легко расщепляются сильными кислотами.
Из группы туйана наиболее широко распространен в различных эфирных маслах (например, в цейлонском кардамоне и майоране) сабинен (D1(7)-туйен). Известны также два изомерных туйона (туйанона-2). Карен (D1(6)-карен) содержится в индийском скипидаре (из сосны длиннохвойной Pinus longifolia); карон (каранон-2) получен синтетически действием щелочи на 8-бромментанон-2 (дигидрокарвонгидробромид).
a-Пинен (D1(6)) и b-пинен (D1(7)) являются главными компонентами скипидара. a-Пинен легко окисляется кислородом воздуха с раскрытием циклобутанового кольца. Разбавленные неорганические кислоты превращают его в терпин (см. разд. IV-2.А.6). Присоединение хлороводорода дает неустойчивый пиненгидрохлорид (1-хлорпинан), который быстро изомеризуется в борнилхлорид (2-хлоркамфан); эта реакция играет ключевую роль в синтезе камфоры. . Циклопропановые кольца в производных карана и туйана и циклобутановое кольцо в производных пинана легко расщепляются сильными кислотами.
Из группы туйана наиболее широко распространен в различных эфирных маслах (например, в цейлонском кардамоне и майоране) сабинен (D1(7)-туйен). Известны также два изомерных туйона (туйанона-2). Карен (D1(6)-карен) содержится в индийском скипидаре (из сосны длиннохвойной Pinus longifolia); карон (каранон-2) получен синтетически действием щелочи на 8-бромментанон-2 (дигидрокарвонгидробромид).
a-Пинен (D1(6)) и b-пинен (D1(7)) являются главными компонентами скипидара. a-Пинен легко окисляется кислородом воздуха с раскрытием циклобутанового кольца. Разбавленные неорганические кислоты превращают его в терпин (см. разд. IV-2.А.6). Присоединение хлороводорода дает неустойчивый пиненгидрохлорид (1-хлорпинан), который быстро изомеризуется в борнилхлорид (2-хлоркамфан); эта реакция играет ключевую роль в синтезе камфоры.
Наиболее важным членом группы камфана является камфора (камфанон-2). Она находится в японском камфарном масле в виде правовращающей формы. Рацемическую форму получают из борнилхлорида обработкой последнего щелочью. Образующийся при этом камфен при гидратации и окислении дает камфору.
 Камфора широко используется в производстве нитроцеллюлозы, целлулоида, бездымного пороха и в медицине. Восстановление камфоры или гидратация камфена дают смесь стереоизомерных спиртов – камфанолов-2 – борнеола и изоборнеола, каждый из которых можно разделить на D- и L-формы. Обе формы борнеола встречаются в природе, например, в Dryobalanops camphora и розмариновом, пихтовом и других маслах.
Декалин и сесквитерпены.
Декалиновая (декагидронафталиновая) система колец также встречается в природных соединениях, в основном в сесквитерпенах, которые содержат 15 атомов углерода (тримеры изопрена).
Камфора широко используется в производстве нитроцеллюлозы, целлулоида, бездымного пороха и в медицине. Восстановление камфоры или гидратация камфена дают смесь стереоизомерных спиртов – камфанолов-2 – борнеола и изоборнеола, каждый из которых можно разделить на D- и L-формы. Обе формы борнеола встречаются в природе, например, в Dryobalanops camphora и розмариновом, пихтовом и других маслах.
Декалин и сесквитерпены.
Декалиновая (декагидронафталиновая) система колец также встречается в природных соединениях, в основном в сесквитерпенах, которые содержат 15 атомов углерода (тримеры изопрена).
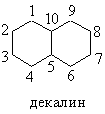 Декалин может существовать в цис- и транс-формах, так как второе кольцо может быть присоединено к первому двумя различными путями, каждый из которых ведет к ненапряженному и неплоскому изомеру. Бициклические сесквитерпены являются диметилизопропилдекалинами с двумя двойными связями. Как и терпены, они построены из изопреновых единиц C5H8. Примером служит кадинен. Кадинен и a-эвдесмол из эвкалиптового масла имеют следующую структуру:
Декалин может существовать в цис- и транс-формах, так как второе кольцо может быть присоединено к первому двумя различными путями, каждый из которых ведет к ненапряженному и неплоскому изомеру. Бициклические сесквитерпены являются диметилизопропилдекалинами с двумя двойными связями. Как и терпены, они построены из изопреновых единиц C5H8. Примером служит кадинен. Кадинен и a-эвдесмол из эвкалиптового масла имеют следующую структуру:
 В. ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Наиболее важными полициклическими соединениями являются стероиды, в основе структуры которых лежит скелет циклопентанопергидрофенантрена. Стероиды входят в состав растительных и животных организмов. К стероидам относятся стерины (например, холестерин), желчные кислоты (например, холевая кислота), половые гормоны (например, эстрон, прогестерон, тестостерон), гормоны коры надпочечников (например, кортикостерон, кортизон), сердечные гликозиды (например, дигитоксин). Трициклические системы встречаются у сесквитерпенов (например, санталена) и дитерпенов (например, абиетиновой кислоты).
В. ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Наиболее важными полициклическими соединениями являются стероиды, в основе структуры которых лежит скелет циклопентанопергидрофенантрена. Стероиды входят в состав растительных и животных организмов. К стероидам относятся стерины (например, холестерин), желчные кислоты (например, холевая кислота), половые гормоны (например, эстрон, прогестерон, тестостерон), гормоны коры надпочечников (например, кортикостерон, кортизон), сердечные гликозиды (например, дигитоксин). Трициклические системы встречаются у сесквитерпенов (например, санталена) и дитерпенов (например, абиетиновой кислоты).
 IV-3. АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Для ароматических соединений характерна ароматичность, т.е. совокупность структурных, энергетических свойств и особенностей реакционной способности циклических структур с системой сопряженных связей. В более узком смысле этот термин относится только к бензоидным соединениями (аренам), в основе структуры которых лежит бензольное кольцо, одно или несколько, в том числе конденсированных, т.е. имеющих два общих атома углерода.
Главные ароматические углеводороды каменноугольной смолы.
Ароматические углеводороды, содержащиеся в каменноугольной смоле, имеют одно или несколько шестичленных колец, которые обычно изображают в структурных формулах с тремя чередующимися двойными связями, – это бензол (т. кип. 80° С), нафталин (т. кип. 218° С, т. пл. 80° С), дифенил (т. кип. 259° С, т. пл. 69° С), флуорен (т. кип. 295° С, т. пл. 114° С), фенантрен (т. кип. 340° С, т. пл. 101° С), антрацен (т. кип. 354° С, т. пл. 216° С), флуорантен (т. пл. 110° С), пирен (т. пл. 151° С), хризен (т. пл. 255° С) (см. также формулы в табл. 4, разд. III).
IV-3. АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ
Для ароматических соединений характерна ароматичность, т.е. совокупность структурных, энергетических свойств и особенностей реакционной способности циклических структур с системой сопряженных связей. В более узком смысле этот термин относится только к бензоидным соединениями (аренам), в основе структуры которых лежит бензольное кольцо, одно или несколько, в том числе конденсированных, т.е. имеющих два общих атома углерода.
Главные ароматические углеводороды каменноугольной смолы.
Ароматические углеводороды, содержащиеся в каменноугольной смоле, имеют одно или несколько шестичленных колец, которые обычно изображают в структурных формулах с тремя чередующимися двойными связями, – это бензол (т. кип. 80° С), нафталин (т. кип. 218° С, т. пл. 80° С), дифенил (т. кип. 259° С, т. пл. 69° С), флуорен (т. кип. 295° С, т. пл. 114° С), фенантрен (т. кип. 340° С, т. пл. 101° С), антрацен (т. кип. 354° С, т. пл. 216° С), флуорантен (т. пл. 110° С), пирен (т. пл. 151° С), хризен (т. пл. 255° С) (см. также формулы в табл. 4, разд. III).
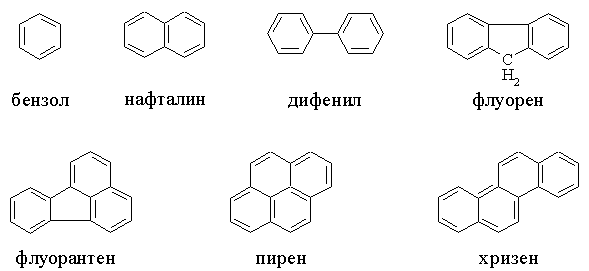 Резонанс в ароматических системах.
На первый взгляд может показаться, что это сильно ненасыщенные соединения, однако двойные связи в них всех, за исключением 9,10-двойной связи фенантрена, исключительно инертны. Это отсутствие реакционной способности или ненормально низкий характер двоесвязности приписывают «резонансу». Резонанс подразумевает, что гипотетические двойные связи не локализованы в специфических или формальных связях. Они делокализованы по всем кольцевым атомам углерода, и невозможно точно изобразить электронную структуру таких молекул единственной формулой обычного типа.
Резонанс в ароматических системах.
На первый взгляд может показаться, что это сильно ненасыщенные соединения, однако двойные связи в них всех, за исключением 9,10-двойной связи фенантрена, исключительно инертны. Это отсутствие реакционной способности или ненормально низкий характер двоесвязности приписывают «резонансу». Резонанс подразумевает, что гипотетические двойные связи не локализованы в специфических или формальных связях. Они делокализованы по всем кольцевым атомам углерода, и невозможно точно изобразить электронную структуру таких молекул единственной формулой обычного типа.
Везде, где возможно написать для молекулы две (или больше) структуры, которые обладают равной или приблизительно равной энергией и которые отличаются только положениями, приписываемыми электронам, обнаруживается, что реальная молекула более стабильна, чем должна была бы быть любая из структур, и обладает свойствами, промежуточными между ними. Приобретенная таким образом дополнительная стабильность называется энергией резонанса. Этот принцип следует из квантовой механики и отражает невозможность точного описания многих из таких микроскопических систем, как атомы и молекулы, простыми схемами.
На основании следующих доказательств можно утверждать, что бензол C6H6 является плоским шестичленным кольцом, содержащим три чередующиеся с простыми двойные связи: гидрирование в жестких условиях превращает его в циклогексан C6H12; озонолиз дает глиоксаль OHC–CHO; дипольные моменты дихлорпроизводных C6H4Cl2 могут быть точно рассчитаны из дипольного момента монохлорбензола, если предположить, что кольцо является плоским правильным шестиугольником. Такой молекуле можно приписать структуру
 Обе эти кекулевские структуры (по имени предложившего их Ф.Кекуле) одинаковы по энергии и делают одинаковый вклад в истинную структуру. Ее можно изобразить как
Обе эти кекулевские структуры (по имени предложившего их Ф.Кекуле) одинаковы по энергии и делают одинаковый вклад в истинную структуру. Ее можно изобразить как  , приписывая полудвоесвязный характер каждой углерод-углеродной связи. Тщательный анализ, проведенный Л.Полингом, показал, что небольшой вклад вносят также дьюаровские структуры: , приписывая полудвоесвязный характер каждой углерод-углеродной связи. Тщательный анализ, проведенный Л.Полингом, показал, что небольшой вклад вносят также дьюаровские структуры:
 Было найдено, что энергия резонанса системы составляет 39 ккал/моль, и, следовательно, бензольная двойная связь стабильнее, чем олефиновая. Поэтому любая реакция, состоящая в присоединении к одной из двойных связей и ведущая к структуре
Было найдено, что энергия резонанса системы составляет 39 ккал/моль, и, следовательно, бензольная двойная связь стабильнее, чем олефиновая. Поэтому любая реакция, состоящая в присоединении к одной из двойных связей и ведущая к структуре  , потребовала бы преодоления высокого энергетического барьера, поскольку две двойные связи в циклогексадиене , потребовала бы преодоления высокого энергетического барьера, поскольку две двойные связи в циклогексадиене  стабилизированы энергией резонанса всего лишь 5 ккал/моль.
Для нафталина можно написать три структуры: стабилизированы энергией резонанса всего лишь 5 ккал/моль.
Для нафталина можно написать три структуры:
 Поскольку все они имеют приблизительно одинаковую энергию, истинная структура является средним арифметическим всех трех и может быть написана как
Поскольку все они имеют приблизительно одинаковую энергию, истинная структура является средним арифметическим всех трех и может быть написана как
 причем дроби указывают степень двоесвязности каждой углерод-углеродной связи. Энергия резонанса составляет 71 ккал/моль. В общем, для бензола пишется только одна кекулевская структура, а первая из написанных выше структур используется для изображения нафталина. Сходным образом изображается структура антрацена (см. табл. 4 в разд. III).
А. АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОЛЬНОГО РЯДА
1. Углеводороды бензольного ряда.
Бензол и его гомологи имеют общую формулу CnH2n – 6. Гомологи состоят из бензольного кольца и одной или нескольких алифатических боковых цепей, присоединенных к его углеродным атомам вместо водорода. Простейший из гомологов – толуол C6H5CH3 – содержится в каменноугольной смоле и имеет существенное значение как исходное соединение для получения взрывчатого вещества тринитротолуола (см. разд. IV-3.А.2 «Нитросоединения») и капролактама. Следующая формула в ряду, C8H10, отвечает четырем соединениям: этилбензолу C6H5C2H5 и ксилолам C6H4(CH3)2. (Высшие гомологи представляют меньший интерес.) Когда к кольцу присоединены два заместителя, возникает возможность изомерии положения; так, существуют три изомерных ксилола:
причем дроби указывают степень двоесвязности каждой углерод-углеродной связи. Энергия резонанса составляет 71 ккал/моль. В общем, для бензола пишется только одна кекулевская структура, а первая из написанных выше структур используется для изображения нафталина. Сходным образом изображается структура антрацена (см. табл. 4 в разд. III).
А. АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ БЕНЗОЛЬНОГО РЯДА
1. Углеводороды бензольного ряда.
Бензол и его гомологи имеют общую формулу CnH2n – 6. Гомологи состоят из бензольного кольца и одной или нескольких алифатических боковых цепей, присоединенных к его углеродным атомам вместо водорода. Простейший из гомологов – толуол C6H5CH3 – содержится в каменноугольной смоле и имеет существенное значение как исходное соединение для получения взрывчатого вещества тринитротолуола (см. разд. IV-3.А.2 «Нитросоединения») и капролактама. Следующая формула в ряду, C8H10, отвечает четырем соединениям: этилбензолу C6H5C2H5 и ксилолам C6H4(CH3)2. (Высшие гомологи представляют меньший интерес.) Когда к кольцу присоединены два заместителя, возникает возможность изомерии положения; так, существуют три изомерных ксилола:
 Другие важные бензольные углеводороды включают ненасыщенный углеводород стирол C6H5CH=CH2, используемый в производстве полимеров; стильбен C6H5CH=CHC6H5; дифенилметан (C6H5)2CH2; трифенилметан (C6H5)3CH; дифенил C6H5–C6H5.
Получение.
Бензольные углеводороды получают следующими методами:
Другие важные бензольные углеводороды включают ненасыщенный углеводород стирол C6H5CH=CH2, используемый в производстве полимеров; стильбен C6H5CH=CHC6H5; дифенилметан (C6H5)2CH2; трифенилметан (C6H5)3CH; дифенил C6H5–C6H5.
Получение.
Бензольные углеводороды получают следующими методами:
1) дегидрогенизация и циклизация парафинов, например:
 2) синтез Вюрца – Фиттига:
2) синтез Вюрца – Фиттига:
 3) реакция Фриделя – Крафтса с алкилгалогенидами или олефинами:
3) реакция Фриделя – Крафтса с алкилгалогенидами или олефинами:
 4) синтез кетонов по Фриделю – Крафтсу с последующим восстановлением по Клемменсену (обработка амальгамой цинка и кислотой), которое превращает карбонильную группу в метиленовое звено:
4) синтез кетонов по Фриделю – Крафтсу с последующим восстановлением по Клемменсену (обработка амальгамой цинка и кислотой), которое превращает карбонильную группу в метиленовое звено:
 5) дегидрогенизация алициклических углеводородов:
5) дегидрогенизация алициклических углеводородов:
 6) декарбоксилирование кислот, например:
6) декарбоксилирование кислот, например:
 7) перегонка фенолов с цинковой пылью (метод полезен для установления структуры, но редко применяется в синтезе) например:
7) перегонка фенолов с цинковой пылью (метод полезен для установления структуры, но редко применяется в синтезе) например:
 Применимы также и другие методы, приведенные выше для получения алифатических углеводородов (например, восстановление галогенидов, спиртов, олефинов).
Реакции
углеводородов бензольного ряда можно подразделить на реакции боковой цепи и реакции кольца. За исключением положения, соседнего с кольцом, боковая цепь ведет себя по существу как парафин, олефин или ацетилен в зависимости от своей структуры. Углерод-водородные связи на углероде, соседнем с кольцом, однако, заметно активированы, особенно по отношению к таким реакциям с участием свободных радикалов, как галогенирование и окисление. Так, толуол и высшие гомологи легко хлорируются и бромируются галогенами на солнечном свету:
Применимы также и другие методы, приведенные выше для получения алифатических углеводородов (например, восстановление галогенидов, спиртов, олефинов).
Реакции
углеводородов бензольного ряда можно подразделить на реакции боковой цепи и реакции кольца. За исключением положения, соседнего с кольцом, боковая цепь ведет себя по существу как парафин, олефин или ацетилен в зависимости от своей структуры. Углерод-водородные связи на углероде, соседнем с кольцом, однако, заметно активированы, особенно по отношению к таким реакциям с участием свободных радикалов, как галогенирование и окисление. Так, толуол и высшие гомологи легко хлорируются и бромируются галогенами на солнечном свету:
 В случае толуола можно ввести второй и третий галогены. Эти a-хлорсоединения легко гидролизуются щелочами:
В случае толуола можно ввести второй и третий галогены. Эти a-хлорсоединения легко гидролизуются щелочами:
 Толуол нетрудно окислить до бензойной кислоты C6H5COOH. Высшие гомологи при окислении претерпевают расщепление боковой цепи до карбоксильной группы, образуя бензойную кислоту.
Главной реакцией кольца является ароматическое замещение, при котором протон замещается положительным атомом или группой, произведенной из кислотного или «электрофильного» реагента:
Толуол нетрудно окислить до бензойной кислоты C6H5COOH. Высшие гомологи при окислении претерпевают расщепление боковой цепи до карбоксильной группы, образуя бензойную кислоту.
Главной реакцией кольца является ароматическое замещение, при котором протон замещается положительным атомом или группой, произведенной из кислотного или «электрофильного» реагента:
 Типичные примеры такого замещения:
а) нитрование, Ar–H + HNO3 ® Ar–NO2 + H2O;
б) галогенирование, Ar–H + X2 ® Ar–X + HX;
в) алкилирование олефинами и алкилгалогенидами по Фриделю – Крафтсу (как указано выше);
Типичные примеры такого замещения:
а) нитрование, Ar–H + HNO3 ® Ar–NO2 + H2O;
б) галогенирование, Ar–H + X2 ® Ar–X + HX;
в) алкилирование олефинами и алкилгалогенидами по Фриделю – Крафтсу (как указано выше);
г) ацилирование по Фриделю – Крафтсу,
 д) сульфирование, Ar–H + H2SO4 (дымящая) ® ArSO3H + H2O.
Введение первого заместителя не встречает осложнений, поскольку все положения в бензоле эквивалентны. Введение второго заместителя происходит в различные положения по отношению к первому заместителю в первую очередь в зависимости от природы группы, уже имеющейся в кольце. Природа атакующего реагента играет второстепенную роль.
д) сульфирование, Ar–H + H2SO4 (дымящая) ® ArSO3H + H2O.
Введение первого заместителя не встречает осложнений, поскольку все положения в бензоле эквивалентны. Введение второго заместителя происходит в различные положения по отношению к первому заместителю в первую очередь в зависимости от природы группы, уже имеющейся в кольце. Природа атакующего реагента играет второстепенную роль.
Группы, которые увеличивают электронную плотность в ароматическом кольце
–O–, –NH2, –N(CH3)2, –OH, –CH3, –OCH3, –NHCOCH3
активируют орто- и пара-положения и направляют следующую группу главным образом в эти позиции.
Напротив, группы, которые оттягивают на себя электроны кольца
 сильнее всего дезактивируют по отношению к электрофильной атаке орто- и пара-положения, поэтому замещение направляется главным образом в мета-положение.
Промежуточными по своему поведению являются некоторые группы, которые благодаря противоположным электронным влияниям дезактивируют кольцо по отношению к дальнейшему замещению, но остаются орто-пара-ориентантами: –Cl, –Br, –I и –CH=CHCOOH. Эти принципы важны для синтеза в ароматическом ряду. Так, чтобы получить п-нитробромбензол
сильнее всего дезактивируют по отношению к электрофильной атаке орто- и пара-положения, поэтому замещение направляется главным образом в мета-положение.
Промежуточными по своему поведению являются некоторые группы, которые благодаря противоположным электронным влияниям дезактивируют кольцо по отношению к дальнейшему замещению, но остаются орто-пара-ориентантами: –Cl, –Br, –I и –CH=CHCOOH. Эти принципы важны для синтеза в ароматическом ряду. Так, чтобы получить п-нитробромбензол  , необходимо сначала бромировать кольцо и затем нитровать его. Обратный порядок операции дает мета-изомер. , необходимо сначала бромировать кольцо и затем нитровать его. Обратный порядок операции дает мета-изомер.
При жестких условиях кольцо можно «заставить» проявить свой скрытый ненасыщенный характер. С очень активными платиновыми катализаторами при давлении водорода в несколько атмосфер можно добиться гидрирования бензола в циклогексан (но никогда не удается получить продукты частичного гидрирования вроде циклогексадиена). Продолжительная обработка хлором и бромом на солнечном свету ведет к образованию гексагалогеноциклогексанов.
2. Замещенные бензола.
Номенклатура.
1) Монозамещенные бензола можно рассматривать как производные бензола, например этилбензол C6H5–C2H5, или как фенилпроизводные алифатических углеводородов, например 2-фенилбутан C6H5–CH(CH3)C2H5, если у них нет тривиальных названий (например, толуол, ксилол). Галогено- и нитропроизводные называют как производные бензола, например нитробензол C6H5NO2, бромбензол C6H5Br. Другие монозамещенные бензола обозначают особыми названиями: фенол C6H5OH, анизол C6H5OCH3, анилин C6H5NH2, бензальдегид C6H5CH=O.
2) В дизамещенных соединениях указывают относительные положения заместителей орто (о), мета (м) и пара (п), как в ксилолах (см. разд. IV-3.А.1). Порядок старшинства в выборе первого заместителя является следующим: COOH, CHO, COR, SO3H, OH, R, NH2, галоген и NO2. Например
 Широко используются некоторые тривиальные названия, например,
Широко используются некоторые тривиальные названия, например,
 3) В случае трех и более заместителей для обозначения положений используются цифры (от 1 до 6). При выборе первого заместителя применяются те же правила старшинства, например:
3) В случае трех и более заместителей для обозначения положений используются цифры (от 1 до 6). При выборе первого заместителя применяются те же правила старшинства, например:
 4) Заместители в боковой цепи: такие соединения обычно называют как арилпроизводные алифатических соединений. Примерами могут служить a-фенилэтиламин (C6H5)CH(NH2)CH3 и a-фенилмасляная кислота C2H5CH(C6H5)COOH. Существуют многочисленные тривиальные названия (например, миндальная кислота C6H5CH(OH)COOH), которые будут рассмотрены при обсуждении соответствующих соединений.
Галогенопроизводные
получают следующими методами:
4) Заместители в боковой цепи: такие соединения обычно называют как арилпроизводные алифатических соединений. Примерами могут служить a-фенилэтиламин (C6H5)CH(NH2)CH3 и a-фенилмасляная кислота C2H5CH(C6H5)COOH. Существуют многочисленные тривиальные названия (например, миндальная кислота C6H5CH(OH)COOH), которые будут рассмотрены при обсуждении соответствующих соединений.
Галогенопроизводные
получают следующими методами:
1) прямое галогенирование кольца
 (Br2 реагирует сходным образом);
2) замещение диазониевой группы (см. ниже «Ароматические амины») галогенид-ионом:
(Br2 реагирует сходным образом);
2) замещение диазониевой группы (см. ниже «Ароматические амины») галогенид-ионом:
 (при X = Cl– и Br– должны быть использованы в качестве катализаторов медь или CuX).
Атомы галогенов в ароматических галогенидах очень инертны к действию оснований. Поэтому реакции замещения, аналогичные реакциям алифатических галогенидов, редко оказываются практически полезными в случае арилгалогенидов. В промышленности гидролиз и аммонолиз хлорбензола достигается в жестких условиях. Замещение нитрогруппой в п- или о-положении активирует галоген по отношению к основаниям. Из бром- и иодбензолов можно приготовить реактив Гриньяра. Хлорбензол не образует реактивов Гриньяра, но из него можно получить фениллитий. Эти ароматические металлоорганические соединения по свойствам похожи на алифатические аналоги.
Нитросоединения
обычно получают прямым нитрованием кольца (см. разд. IV-3.А.1, «Реакции») смесью концентрированных азотной и серной кислот. Реже их готовят окислением нитрозосоединений (C6H5NO). Введение одной нитрогруппы в бензол протекает относительно просто. Вторая входит более медленно. Третью удается ввести только при продолжительной обработке смесью дымящих азотной и серной кислот. Это общий эффект м-ориентирующих групп; они всегда снижают способность кольца к дальнейшему замещению. Тринитробензолы ценятся как взрывчатые вещества. Для осуществления их синтеза нитрование обычно проводят не на самом бензоле, а на таких его производных, как толуол или фенол, в которых о,п-ориентирующие заместители могут активировать кольцо. Общеизвестными примерами служат 2,4,6-тринитрофенол (пикриновая кислота) и 2,4,6-тринитротолуол (тротил).
(при X = Cl– и Br– должны быть использованы в качестве катализаторов медь или CuX).
Атомы галогенов в ароматических галогенидах очень инертны к действию оснований. Поэтому реакции замещения, аналогичные реакциям алифатических галогенидов, редко оказываются практически полезными в случае арилгалогенидов. В промышленности гидролиз и аммонолиз хлорбензола достигается в жестких условиях. Замещение нитрогруппой в п- или о-положении активирует галоген по отношению к основаниям. Из бром- и иодбензолов можно приготовить реактив Гриньяра. Хлорбензол не образует реактивов Гриньяра, но из него можно получить фениллитий. Эти ароматические металлоорганические соединения по свойствам похожи на алифатические аналоги.
Нитросоединения
обычно получают прямым нитрованием кольца (см. разд. IV-3.А.1, «Реакции») смесью концентрированных азотной и серной кислот. Реже их готовят окислением нитрозосоединений (C6H5NO). Введение одной нитрогруппы в бензол протекает относительно просто. Вторая входит более медленно. Третью удается ввести только при продолжительной обработке смесью дымящих азотной и серной кислот. Это общий эффект м-ориентирующих групп; они всегда снижают способность кольца к дальнейшему замещению. Тринитробензолы ценятся как взрывчатые вещества. Для осуществления их синтеза нитрование обычно проводят не на самом бензоле, а на таких его производных, как толуол или фенол, в которых о,п-ориентирующие заместители могут активировать кольцо. Общеизвестными примерами служат 2,4,6-тринитрофенол (пикриновая кислота) и 2,4,6-тринитротолуол (тротил).
Единственные полезные реакции нитросоединений – это реакции их восстановления. Сильные восстанавливающие агенты (активированный катализаторами водород, олово и соляная кислота, бисульфид-ион) превращают их прямо в амины. Контролируемое электролитическое восстановление позволяет выделить следующие промежуточные стадии:
 Бисульфид аммония является специфическим реагентом для превращения динитросоединений в нитроанилины, например:
Бисульфид аммония является специфическим реагентом для превращения динитросоединений в нитроанилины, например:
 Ароматические амины.
Первичные амины получают восстановлением соответствующих нитросоединений. Они представляют собой очень слабые основания (K = 10–10). N-алкиланилины можно приготовить алкилированием первичных аминов. Они по большинству реакций напоминают алифатические амины, за исключением взаимодействия с азотистой кислотой и с окислителями.
Ароматические амины.
Первичные амины получают восстановлением соответствующих нитросоединений. Они представляют собой очень слабые основания (K = 10–10). N-алкиланилины можно приготовить алкилированием первичных аминов. Они по большинству реакций напоминают алифатические амины, за исключением взаимодействия с азотистой кислотой и с окислителями.
С азотистой кислотой в кислой среде (при 0–5 ° С) первичные амины дают устойчивые соли диазония (C6H5N=N+X–), имеющие много важных синтетических приложений. Замещение диазониевой группы галогеном уже обсуждалось. Эта группа может быть также заменена на цианид-ион (с CuCN в качестве катализатора) с получением ароматических нитрилов (C6H5CN). Кипящая вода превращает соли диазония в фенолы. В кипящем спирте эта группа замещается на водород:
 В почти нейтральном растворе диазониевые соли сочетаются с фенолами (и многими аминами), давая азокрасители:
В почти нейтральном растворе диазониевые соли сочетаются с фенолами (и многими аминами), давая азокрасители:
 Эта реакция имеет огромное значение для промышленности синтетических красителей. Восстановление бисульфитом ведет к арилгидразинам C6H5NHNH2.
Вторичные ариламины, подобно алифатическим вторичным аминам, дают N-нитрозосоединения. Третичные ариламины C6H5NRRў, однако, дают п-нитрозоариламины (например, п-ON–C6H4NRRў). Эти соединения имеют некоторое значение для приготовления чистых вторичных алифатических аминов, поскольку они легко гидролизуются до вторичного амина RRўNH и п-нитрозофенола.
Эта реакция имеет огромное значение для промышленности синтетических красителей. Восстановление бисульфитом ведет к арилгидразинам C6H5NHNH2.
Вторичные ариламины, подобно алифатическим вторичным аминам, дают N-нитрозосоединения. Третичные ариламины C6H5NRRў, однако, дают п-нитрозоариламины (например, п-ON–C6H4NRRў). Эти соединения имеют некоторое значение для приготовления чистых вторичных алифатических аминов, поскольку они легко гидролизуются до вторичного амина RRўNH и п-нитрозофенола.
Окисление ароматических аминов может затрагивать не только аминогруппу, но и п-положение кольца. Так, анилин при окислении превращается во множество продуктов, включая азобензол, нитробензол, хинон ( и анилиновый черный краситель).
Арилалкиламины (например, бензиламин C6H5CH2NH2) обнаруживают те же свойства и реакции, что и алкиламины с той же молекулярной массой.
Фенолы,
ароматические гидроксисоединения, у которых гидроксильная группа присоединена прямо к кольцу. Они значительно более кислотны, чем спирты, располагаясь по силе между угольной кислотой и бикарбонат-ионом (для фенола Ka = 10–10). Наиболее общий метод их получения – разложение солей диазония. Их соли можно получить сплавлением солей арилсульфокислот со щелочью: и анилиновый черный краситель).
Арилалкиламины (например, бензиламин C6H5CH2NH2) обнаруживают те же свойства и реакции, что и алкиламины с той же молекулярной массой.
Фенолы,
ароматические гидроксисоединения, у которых гидроксильная группа присоединена прямо к кольцу. Они значительно более кислотны, чем спирты, располагаясь по силе между угольной кислотой и бикарбонат-ионом (для фенола Ka = 10–10). Наиболее общий метод их получения – разложение солей диазония. Их соли можно получить сплавлением солей арилсульфокислот со щелочью:
 В дополнение к этим методам фенол получают в промышленности прямым окислением бензола и гидролизом хлорбензола в жестких условиях – раствором едкого натра при высокой температуре под давлением. Фенол и некоторые из его простейших гомологов – метилфенолы (крезолы) и диметилфенолы (ксиленолы) – найдены в каменноугольной смоле.
Реакции фенолов примечательны лабильностью гидроксильного водорода и устойчивостью гидроксильной группы к замещению. Кроме того, пара-положение (и орто-положения, если пара-положение блокировано) очень чувствительны к атаке реагентов, вызывающих ароматическое замещение, и окислителей. Фенолы легко образуют натриевые соли при обработке едким натром и содой, но не бикарбонатом натрия. Эти соли легко реагируют с ангидридом и хлорангидридом кислот, давая сложные эфиры (например, C6H5OOCCH3), и с алкилгалогенидами и алкилсульфатами, образуя простые эфиры (например, анизол C6H5OCH3). Сложные эфиры фенолов можно также получить действием ацилирующих агентов в присутствии пиридина. Фенольные гидроксильные группы можно удалить перегонкой фенолов с цинковой пылью, но они не замещаются при нагревании с галогеноводородными кислотами, как спиртовые гидроксильные группы.
В дополнение к этим методам фенол получают в промышленности прямым окислением бензола и гидролизом хлорбензола в жестких условиях – раствором едкого натра при высокой температуре под давлением. Фенол и некоторые из его простейших гомологов – метилфенолы (крезолы) и диметилфенолы (ксиленолы) – найдены в каменноугольной смоле.
Реакции фенолов примечательны лабильностью гидроксильного водорода и устойчивостью гидроксильной группы к замещению. Кроме того, пара-положение (и орто-положения, если пара-положение блокировано) очень чувствительны к атаке реагентов, вызывающих ароматическое замещение, и окислителей. Фенолы легко образуют натриевые соли при обработке едким натром и содой, но не бикарбонатом натрия. Эти соли легко реагируют с ангидридом и хлорангидридом кислот, давая сложные эфиры (например, C6H5OOCCH3), и с алкилгалогенидами и алкилсульфатами, образуя простые эфиры (например, анизол C6H5OCH3). Сложные эфиры фенолов можно также получить действием ацилирующих агентов в присутствии пиридина. Фенольные гидроксильные группы можно удалить перегонкой фенолов с цинковой пылью, но они не замещаются при нагревании с галогеноводородными кислотами, как спиртовые гидроксильные группы.
Гидроксильная группа так сильно активирует орто- и пара-положения, что реакции нитрования, сульфирования, галогенирования и им подобные протекают бурно даже при низких температурах. Действие бромной воды на фенол ведет к 2,4,6-трибромфенолу, но п-бромфенол можно получить бромированием в таких растворителях, как сероуглерод, при низких температурах. Галогенирование без растворителя дает смесь о- и п-галогенофенолов. Разбавленная азотная кислота легко нитрует фенол, давая смесь о- и п-нитрофенолов, из которой о-нитрофенол можно отогнать с паром.
Фенол и крезолы используют как дезинфицирующие средства. Среди других фенолов важное значение имеют:
а) карвакрол (2-метил-5-изопропилфенол) и тимол (3-метил-6-изопропилфенол), которые встречаются во многих эфирных маслах как продукты химических превращений терпенов;
б) анол ( п-пропенилфенол), который встречается в виде соответствующего метилового эфира анетола в анисовом масле; близкий к нему хавикол (п-аллилфенол) находится в маслах из листьев бетеля и лавра и в виде метилового эфира, эстрагола, в анисовом масле;
в) пирокатехин (2-гидроксифенол), который встречается во многих растениях; в промышленности его получают гидролизом (в жестких условиях) о-дихлорбензола или о-хлорфенола, а также деметилированием гваякола (монометилового эфира пирокатехина), содержащегося в продуктах сухой перегонки бука; пирокатехин легко окисляется в о-хинон
 и находит широкое применение как восстановитель в фотографических проявителях;
г) резорцин (м-гидроксифенол); его получают щелочным плавлением м-бензолдисульфокислоты и используют для приготовления красителей; он легко замещается в положении 4 и восстанавливается в дигидрорезорцин (циклогександион-1,3), который расщепляется разбавленной щелочью в d-кетокапроновую кислоту; его 4-н-гексилпроизводное является полезным антисептиком;
и находит широкое применение как восстановитель в фотографических проявителях;
г) резорцин (м-гидроксифенол); его получают щелочным плавлением м-бензолдисульфокислоты и используют для приготовления красителей; он легко замещается в положении 4 и восстанавливается в дигидрорезорцин (циклогександион-1,3), который расщепляется разбавленной щелочью в d-кетокапроновую кислоту; его 4-н-гексилпроизводное является полезным антисептиком;
д) гидрохинон ( п-оксифенол), который встречается в некоторых растениях в виде гликозида арбутина; его получают восстановлением хинона (см. выше «Ароматические амины»), продукта окисления анилина; это легко обратимая реакция; при 50%-ном ее протекании образуется устойчивое эквимолекулярное соединение хинона и гидрохинона – хингидрон; хингидронный электрод часто применяется в потенциометрическом анализе; благодаря восстанавливающим свойствам гидрохинона он, подобно пирокатехину, используется в фотографических проявителях;
е) пирогаллол (2,3-дигидроксифенол), который получают из галловой кислоты ( см. ниже «Ароматические кислоты») перегонкой над пемзой в атмосфере углекислого газа; будучи мощным восстановителем, пирогаллол находит применение как поглотитель кислорода в газовом анализе и как фотографический проявитель.
Ароматические спирты
– это соединения, которые, подобно бензиловому спирту C6H5CH2OH, содержат гидроксильную группу в боковой цепи (а не в кольце, как фенолы). Если гидроксильная группа находится при углеродном атоме, соседнем с кольцом, она особенно легко замещается на галоген при действии галогеноводородов на водород (над платиной) и легко отщепляется при дегидратации (в C6H5CHOHR). Такие простые ароматические спирты, как бензиловый, фенетиловый (C6H5CH2CH2OH), фенилпропиловый (C6H5CH2CH2CH2OH) и коричный (C6H5CH=CHCH2OH), используются в парфюмерной промышленности и встречаются в природе во многих эфирных маслах. Их можно получить по любой из общих реакций, описанных выше для приготовления алифатических спиртов.
Ароматические альдегиды.
Бензальдегид C6H5CHO, простейший ароматический альдегид, образуется в масле горького миндаля в результате ферментативного гидролиза гликозида амигдалина C6H5CH(CN)–O–C12H21O10. Он находит широкое применение как промежуточное вещество в синтезе красителей и других ароматических соединений, а также как отдушка и основа духов. В промышленности его получают гидролизом бензилиденхлорида C6H5CHCl2, продукта хлорирования толуола, или прямым окислением толуола в газовой (над V2O5) либо в жидкой фазе с MnO2 в 65%-ной серной кислоте при 40° С.
Для приготовления ароматических альдегидов служат следующие общие методы:
1) синтез Гаттермана – Коха:
 2) синтез Гаттермана:
2) синтез Гаттермана:
 3) синтез Раймера – Тимана (для получения ароматических гидроксиальдегидов):
3) синтез Раймера – Тимана (для получения ароматических гидроксиальдегидов):
 Бензальдегид окисляется кислородом воздуха в бензойную кислоту; этого можно достичь также применением других окислителей, например перманганата или дихромата. Вообще бензальдегид и другие ароматические альдегиды вступают в карбонильные реакции конденсации (см. разд. IV-1.А.4) несколько менее активно, чем алифатические альдегиды. Отсутствие a-водородного атома препятствует вступлению ароматических альдегидов в альдольную самоконденсацию. Тем не менее смешанная альдольная конденсация используется в синтезе:
Бензальдегид окисляется кислородом воздуха в бензойную кислоту; этого можно достичь также применением других окислителей, например перманганата или дихромата. Вообще бензальдегид и другие ароматические альдегиды вступают в карбонильные реакции конденсации (см. разд. IV-1.А.4) несколько менее активно, чем алифатические альдегиды. Отсутствие a-водородного атома препятствует вступлению ароматических альдегидов в альдольную самоконденсацию. Тем не менее смешанная альдольная конденсация используется в синтезе:
 Следующие реакции типичны для ароматических альдегидов:
Следующие реакции типичны для ароматических альдегидов:
1) реакция Канниццаро:
 2) бензоиновая конденсация:
2) бензоиновая конденсация:
 3) реакция Перкина:
3) реакция Перкина:
 Некоторое значение имеют следующие ароматические альдегиды:
Некоторое значение имеют следующие ароматические альдегиды:
1) Салициловый альдегид ( о-гидроксибензальдегид) встречается в природе в душистом масле таволги. Его получают из фенола синтезом Раймера – Тимана. Он находит применение в синтезе кумарина (см. разд. IV-4.Г) и некоторых красителей.
2) Коричный альдегид C6H5CH=CHCHO содержится в масле корицы и кассии. Его получают кротоновой конденсацией (см. разд. IV-1.А.4) бензальдегида с уксусным альдегидом.
3) Анисовый альдегид ( п-метоксибензальдегид) содержится в масле кассии и используется в духах и ароматизирующих добавках. Его получают синтезом Гаттермана из анизола.
4) Ванилин (3-метокси-4-гидроксибензальдегид) является главным ароматическим компонентом экстрактов ванили. Его можно получить по реакции Раймера – Тимана из гваякола или обработкой эвгенола (2-метокси-4-аллилфенола) щелочью с последующим окислением.
5) Пиперональ обладает запахом гелиотропа. Его получают из сафрола (масло американского лавра) аналогично тому, как получают ванилин из эвгенола.
 Ароматические кетоны.
Эти вещества обычно получают из ароматических соединений и хлорангидридов кислот по реакции Фриделя – Крафтса. Применяются также общие методы получения алифатических кетонов. Специфическим методом получения гидроксикетонов является перегруппировка Фриса в сложных эфирах фенолов:
Ароматические кетоны.
Эти вещества обычно получают из ароматических соединений и хлорангидридов кислот по реакции Фриделя – Крафтса. Применяются также общие методы получения алифатических кетонов. Специфическим методом получения гидроксикетонов является перегруппировка Фриса в сложных эфирах фенолов:
 (при повышенных температурах порядка 165–170° С преобладает о-изомер). Вообще ароматические кетоны вступают в те же реакции, что и алифатические кетоны, но гораздо более медленно. a-Дикетонбензил C6H5CO–COC6H5, получаемый окислением бензоина (см. предыдущий разд. «Ароматические альдегиды»), претерпевает характерную перегруппировку при обработке щелочью, образуя бензиловую кислоту (C6H5)2C(OH)COOH.
Ароматические кислоты.
Простейшей ароматической карбоновой кислотой является бензойная C6H5COOH, которая вместе с ее эфирами встречается в природе в составе многих смол и бальзамов. Она широко применяется как пищевой консервант, особенно в форме натриевой соли. Как и алифатические кислоты, бензойную кислоту и другие ароматические кислоты можно получить действием углекислого газа на реактив Гриньяра (например, C6H5MgBr). Их можно также приготовить гидролизом соответствующих нитрилов, которые в ароматическом ряду получают из диазониевых солей или сплавлением натриевых солей ароматических сульфокислот с цианидом натрия:
(при повышенных температурах порядка 165–170° С преобладает о-изомер). Вообще ароматические кетоны вступают в те же реакции, что и алифатические кетоны, но гораздо более медленно. a-Дикетонбензил C6H5CO–COC6H5, получаемый окислением бензоина (см. предыдущий разд. «Ароматические альдегиды»), претерпевает характерную перегруппировку при обработке щелочью, образуя бензиловую кислоту (C6H5)2C(OH)COOH.
Ароматические кислоты.
Простейшей ароматической карбоновой кислотой является бензойная C6H5COOH, которая вместе с ее эфирами встречается в природе в составе многих смол и бальзамов. Она широко применяется как пищевой консервант, особенно в форме натриевой соли. Как и алифатические кислоты, бензойную кислоту и другие ароматические кислоты можно получить действием углекислого газа на реактив Гриньяра (например, C6H5MgBr). Их можно также приготовить гидролизом соответствующих нитрилов, которые в ароматическом ряду получают из диазониевых солей или сплавлением натриевых солей ароматических сульфокислот с цианидом натрия:
 Другие методы их получения включают:
Другие методы их получения включают:
1) окислительное расщепление алифатических боковых цепей
 2) гидролиз трихлорметиларенов
2) гидролиз трихлорметиларенов
 3) синтез гидроксикислот по Кольбе
3) синтез гидроксикислот по Кольбе
 4) окисление ацетофенонов гипогалогенитами
4) окисление ацетофенонов гипогалогенитами
 Ниже перечислены некоторые из наиболее важных ароматических карбоновых кислот:
1) Салициловую (о-гидроксибензойную) кислоту о-C6H4(COOH)OH получают из фенола синтезом Кольбе. Ее метиловый эфир является душистым компонентом масла зимолюбки (гаультерии), а натриевая соль ацетилпроизводного представляет собой аспирин (о-ацетоксибензоат натрия).
Ниже перечислены некоторые из наиболее важных ароматических карбоновых кислот:
1) Салициловую (о-гидроксибензойную) кислоту о-C6H4(COOH)OH получают из фенола синтезом Кольбе. Ее метиловый эфир является душистым компонентом масла зимолюбки (гаультерии), а натриевая соль ацетилпроизводного представляет собой аспирин (о-ацетоксибензоат натрия).
2) Фталевую ( о-карбоксибензойную) кислоту получают окислением нафталина. Она легко образует ангидрид, а последний при действии аммиака дает фталимид – важное промежуточное вещество в синтезе многих соединений, включая краситель индиго
 3) Антраниловая (о-аминобензойная) кислота о-C6H4(NH2)COOH получается при действии гипохлорита натрия на фталимид (реакция Гофмана). Ее метиловый эфир является компонентом духов и встречается в природе в маслах жасмина и апельсиновых листьев.
3) Антраниловая (о-аминобензойная) кислота о-C6H4(NH2)COOH получается при действии гипохлорита натрия на фталимид (реакция Гофмана). Ее метиловый эфир является компонентом духов и встречается в природе в маслах жасмина и апельсиновых листьев.
4) Галловая (3,4,5-тригидроксибензойная) кислота образуется вместе с глюкозой при гидролизе некоторых сложных веществ растительного происхождения, известных под названием таннинов.
Сульфокислоты.
Бензолсульфокислота C6H5SO3H получается при действии дымящей серной кислоты на бензол. Она и другие сульфокислоты являются сильными кислотами (K > 0,1). Сульфокислоты легко растворимы в воде, гигроскопичны; их трудно получить в свободном состоянии. Чаще всего их используют в виде натриевых солей.
Наиболее важные реакции солей, а именно сплавление со щелочами (с образованием фенолов) и с цианидом натрия (с получением нитрилов), уже обсуждались. При действии пентахлорида фосфора они дают арилсульфохлориды (например, C6H5SO2Cl), которые находят применение в алифатических и алициклических синтезах. Наиболее часто используемым таким способом арилсульфохлоридом является п-толуолсульфохлорид (п-CH3C6H4SO2Cl), в литературе часто называемый тозилхлоридом (TsCl). Нагревание сульфокислот в 50–60% серной кислоте при 150° С вызывает их гидролиз до серной кислоты и исходных углеводородов:
 Важной сульфокислотой является сульфаниловая кислота п-H2NC6H4SO3H (или п-H3N+C6H4SO3–), амид (сульфаниламид) и другие производные которой представляют собой важные химиотерапевтические средства. Сульфаниловую кислоту получают действием дымящей серной кислоты на анилин.
Многие моющие средства являются солями длинноцепочечных сульфокислот, например NaO3S–C6H4–C12H25.
Б. АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ НАФТАЛИНОВОГО РЯДА
1. Синтез a- и b-замещенных производных нафталина.
Нафталин является главным компонентом каменноугольной смолы. Он имеет исключительное значение в синтезе многих промышленных продуктов, включая индиго и азокрасители. Однако его использование как репеллента, отпугивающего моль, сократилось с введением таких новых средств, как п-дихлорбензол. Его монозамещенные производные обозначаются как a- или b- в соответствии с положением заместителя (см. табл. 4 в разд. III). Положения в полизамещенных производных обозначаются цифрами.
Важной сульфокислотой является сульфаниловая кислота п-H2NC6H4SO3H (или п-H3N+C6H4SO3–), амид (сульфаниламид) и другие производные которой представляют собой важные химиотерапевтические средства. Сульфаниловую кислоту получают действием дымящей серной кислоты на анилин.
Многие моющие средства являются солями длинноцепочечных сульфокислот, например NaO3S–C6H4–C12H25.
Б. АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ НАФТАЛИНОВОГО РЯДА
1. Синтез a- и b-замещенных производных нафталина.
Нафталин является главным компонентом каменноугольной смолы. Он имеет исключительное значение в синтезе многих промышленных продуктов, включая индиго и азокрасители. Однако его использование как репеллента, отпугивающего моль, сократилось с введением таких новых средств, как п-дихлорбензол. Его монозамещенные производные обозначаются как a- или b- в соответствии с положением заместителя (см. табл. 4 в разд. III). Положения в полизамещенных производных обозначаются цифрами.
Вообще говоря, a-положение проявляет более высокую реакционную способность. Нитрование, галогенирование и низкотемпературное сульфирование ведут к a-производным. Доступ к b-положению достигается главным образом посредством высокотемпературного сульфирования. В этих условиях a-сульфокислота перегруппировывается в более устойчивую b-форму. Введение других заместителей в b-положение становится после этого возможным при помощи реакции Бухерера: сначала из b-нафталинсульфокислоты щелочным плавлением получают b-нафтол b-C10H7OH, который затем при обработке бисульфитом аммония при 150° С и 6 атм дает b-нафтиламин b-C10H7NH2; через соединения диазония, получаемые из этого амина обычным путем, теперь можно ввести в b-положение галоген или цианогруппу. Реакция Фриделя – Крафтса между нафталином и хлорангидридом также дает b-ацилпроизводные b-C10H7COR.
2. Реакции замещения производных нафталина.
Реакции производных нафталина те же, что и реакции производных бензола. Так, нафталинсульфокислоты служат источником нафтолов; нафтиламины через соли диазония превращаются в галогено- и цианнафталины. Поэтому особое обсуждение реакций соединений нафталина будет опущено.
Однако реакции замещения в производных нафталина представляют определенный интерес.
1) При наличии о,п-ориентанта (–CH3, –OH) в 1(a)-положении атака направляется преимущественно в положение 4 и затем в положение 2.
2) В присутствии м-ориентанта (–NO2) в положении 1 атака направляется в положение 8 (пери) и затем в положение 5.
3) При наличии о,п-ориентанта в положении 2 (b) атаке подвергается преимущественно положение 1, хотя сульфирование может происходить в положении 6. Особенно важно, что никогда не подвергается атаке положение 3. Это объясняют низкой степенью двоесвязности углерод-углеродной связи 2-3.
В нафталине замещение протекает в более мягких условиях, чем в бензоле. Нафталин также легче восстанавливается. Так, амальгама натрия восстанавливает его в тетралин (тетрагидронафталин; формулу см. в табл. 4 в разд. III). Он также более чувствителен к окислению. Горячая концентрированная серная кислота в присутствии ионов ртути превращает его во фталевую кислоту (см. разд. IV-3.А.2 «Ароматические кислоты»). Хотя в толуоле метильная группа окисляется раньше кольца, в b-метилнафталине положения 1,4 более подвержены окислению, так что первым продуктом является 2-метил-1,4-нафтохинон:
 В. ПРОИЗВОДНЫЕ МНОГОЯДЕРНЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
1. Антрацен и его производные.
Антрацен (формулу см. в табл. 4, разд. III) содержится в значительных количествах в каменноугольной смоле и находит широкое применение в промышленности как промежуточное вещество в синтезе красителей. Положения 9,10 высокореакционноспособны в реакциях присоединения. Так, водород и бром легко присоединяются, давая соответственно 9,10-дигидро- и 9,10-дибромантрацен. Окисление хромовой кислотой превращает антрацен в антрахинон.
В. ПРОИЗВОДНЫЕ МНОГОЯДЕРНЫХ АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ
1. Антрацен и его производные.
Антрацен (формулу см. в табл. 4, разд. III) содержится в значительных количествах в каменноугольной смоле и находит широкое применение в промышленности как промежуточное вещество в синтезе красителей. Положения 9,10 высокореакционноспособны в реакциях присоединения. Так, водород и бром легко присоединяются, давая соответственно 9,10-дигидро- и 9,10-дибромантрацен. Окисление хромовой кислотой превращает антрацен в антрахинон.
 Антрахинон (т. пл. 285° С) представляет собой желтое кристаллическое вещество. Наиболее общий способ получения антрахинона и его производных состоит в циклизации о-бензоилбензойных кислот при действии серной кислоты
Антрахинон (т. пл. 285° С) представляет собой желтое кристаллическое вещество. Наиболее общий способ получения антрахинона и его производных состоит в циклизации о-бензоилбензойных кислот при действии серной кислоты
 о-Бензоилбензойные кислоты получают действием фталевого ангидрида на бензол (или соответствующее его производное) в присутствии хлорида алюминия. Антрахинон чрезвычайно устойчив к окислению. Такие восстановители, как цинковая пыль и щелочь или бисульфит натрия, превращают его в антрагидрохинон (9,10-дигидроксиантрацен), белое вещество, растворяющееся в щелочи с образованием кроваво-красных растворов. Олово и соляная кислота восстанавливают одну кетогруппу в метиленовую, образуя антрон. Нитрование в жестких условиях дает главным образом a(1)-производное вместе с заметным количеством 1,5- и 1,8-динитроантрахинонов. Сульфирование серной кислотой приводит к образованию главным образом b(2)-сульфокислоты, но в присутствии небольших количеств сульфата ртути основным продуктом является a-сульфокислота. Дисульфирование в присутствии сульфата ртути дает в основном 1,5- и 1,8-дисульфокислоты. В отсутствие ртути образуются 2,6- и 2,7-дисульфокислоты. Сульфокислоты антрахинона имеют большое значение, так как из них щелочным плавлением получают гидроксиантрахиноны, многие из которых являются ценными красителями. Так, окислительное щелочное плавление b-сульфокислоты дает краситель ализарин (1,2-дигидроксиантрахинон), который в природе содержится в корнях марены. Сульфокислотные группы в антрахиноне можно также непосредственно заменить аминогруппами с образованием аминоантрахинонов, представляющих собой ценные красители. В этой реакции натриевую соль сульфокислоты обрабатывают аммиаком при 175–200° С в присутствии мягкого окислителя (например, мышьяковокислого натрия), добавляемого, чтобы разрушить образующийся сульфит.
2. Фенантрен и его производные.
В природе фенантрен находится в каменноугольной смоле. Он сам и его производные могут быть получены из о-нитростильбенкарбоновой кислоты, образующейся конденсацией о-нитробензальдегида и фенилуксусной кислоты по методу Пшорра:
о-Бензоилбензойные кислоты получают действием фталевого ангидрида на бензол (или соответствующее его производное) в присутствии хлорида алюминия. Антрахинон чрезвычайно устойчив к окислению. Такие восстановители, как цинковая пыль и щелочь или бисульфит натрия, превращают его в антрагидрохинон (9,10-дигидроксиантрацен), белое вещество, растворяющееся в щелочи с образованием кроваво-красных растворов. Олово и соляная кислота восстанавливают одну кетогруппу в метиленовую, образуя антрон. Нитрование в жестких условиях дает главным образом a(1)-производное вместе с заметным количеством 1,5- и 1,8-динитроантрахинонов. Сульфирование серной кислотой приводит к образованию главным образом b(2)-сульфокислоты, но в присутствии небольших количеств сульфата ртути основным продуктом является a-сульфокислота. Дисульфирование в присутствии сульфата ртути дает в основном 1,5- и 1,8-дисульфокислоты. В отсутствие ртути образуются 2,6- и 2,7-дисульфокислоты. Сульфокислоты антрахинона имеют большое значение, так как из них щелочным плавлением получают гидроксиантрахиноны, многие из которых являются ценными красителями. Так, окислительное щелочное плавление b-сульфокислоты дает краситель ализарин (1,2-дигидроксиантрахинон), который в природе содержится в корнях марены. Сульфокислотные группы в антрахиноне можно также непосредственно заменить аминогруппами с образованием аминоантрахинонов, представляющих собой ценные красители. В этой реакции натриевую соль сульфокислоты обрабатывают аммиаком при 175–200° С в присутствии мягкого окислителя (например, мышьяковокислого натрия), добавляемого, чтобы разрушить образующийся сульфит.
2. Фенантрен и его производные.
В природе фенантрен находится в каменноугольной смоле. Он сам и его производные могут быть получены из о-нитростильбенкарбоновой кислоты, образующейся конденсацией о-нитробензальдегида и фенилуксусной кислоты по методу Пшорра:
 Двойная связь в положении 9,10 высокореакционноспособна; она легко присоединяет бром и водород и претерпевает окисление сначала в 9,10-фенантрахинон, а затем до дифеновой кислоты
Двойная связь в положении 9,10 высокореакционноспособна; она легко присоединяет бром и водород и претерпевает окисление сначала в 9,10-фенантрахинон, а затем до дифеновой кислоты
 Реакции замещения в фенантрене обычно идут в положения 2, 3, 6 и 7.
3. Высшие многоядерные углеводороды
привлекают к себе внимание главным образом в связи с их высокой канцерогенной активностью. Вот некоторые примеры:
Реакции замещения в фенантрене обычно идут в положения 2, 3, 6 и 7.
3. Высшие многоядерные углеводороды
привлекают к себе внимание главным образом в связи с их высокой канцерогенной активностью. Вот некоторые примеры:
 Красители пирантрон, идантреновый желтый и виолантрон являются кетопроизводными сложных многоядерных углеводородов.
IV-4. ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Гетероциклические соединения – это углеродные циклические соединения, в которых один или несколько атомов кольцевой системы являются отличными от углерода неметаллами (кислородом, азотом или серой). Как и карбоциклические соединения, гетероциклы можно подразделить на имеющие ароматический характер и продукты восстановления таких ароматических гетероциклов, которые аналогично алициклическим соединениям обнаруживают свойства и реакции, сходные со свойствами и реакциями алифатических соединений.
Красители пирантрон, идантреновый желтый и виолантрон являются кетопроизводными сложных многоядерных углеводородов.
IV-4. ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Гетероциклические соединения – это углеродные циклические соединения, в которых один или несколько атомов кольцевой системы являются отличными от углерода неметаллами (кислородом, азотом или серой). Как и карбоциклические соединения, гетероциклы можно подразделить на имеющие ароматический характер и продукты восстановления таких ароматических гетероциклов, которые аналогично алициклическим соединениям обнаруживают свойства и реакции, сходные со свойствами и реакциями алифатических соединений.
Гетероциклы удобно классифицировать а) по числу атомов в кольце, б) по числу и природе гетероатомов. Ненасыщенные гетероциклы, обнаруживающие максимально ароматический характер, берутся в качестве ключевых представителей каждой циклической системы.
А. ПЯТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
1. Один гетероатом
 2. Два гетероатома
2. Два гетероатома
 3. Три и более гетероатомов
3. Три и более гетероатомов
 Резонанс (см. «Резонанс» в начале разд. IV-3) пятичленных колец включает значительный вклад следующих структур:
Резонанс (см. «Резонанс» в начале разд. IV-3) пятичленных колец включает значительный вклад следующих структур:
 Приобретенная таким путем энергия резонанса делает эти системы весьма устойчивыми к реакциям присоединения по двойным связям, и они вступают во многие типичные реакции ароматического замещения.
Б. ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
1. Один гетероатом
Приобретенная таким путем энергия резонанса делает эти системы весьма устойчивыми к реакциям присоединения по двойным связям, и они вступают во многие типичные реакции ароматического замещения.
Б. ШЕСТИЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ
1. Один гетероатом
 2. Два гетероатома
2. Два гетероатома
 В. КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СИСТЕМЫ
Важные ряды соединений в каждом классе получают конденсацией гетероциклического кольца с одним или несколькими бензольными, например:
В. КОНДЕНСИРОВАННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СИСТЕМЫ
Важные ряды соединений в каждом классе получают конденсацией гетероциклического кольца с одним или несколькими бензольными, например:
 Гетероциклические системы широко распространены в природе, особенно в алкалоидах, растительных пигментах (антоцианины, флавоны), порфиринах (гемин, хлорофилл) и витаминах группы В (тиамин, рибофлавин, фолевая кислота).
Гетероциклические системы широко распространены в природе, особенно в алкалоидах, растительных пигментах (антоцианины, флавоны), порфиринах (гемин, хлорофилл) и витаминах группы В (тиамин, рибофлавин, фолевая кислота).
Ниже рассмотрены подробнее некоторые гетероциклические соединения.
Г. ПРАКТИЧЕСКИ ВАЖНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ
Фуран,
летучая жидкость, устойчивая к действию щелочей, но чувствительная к кислотам
 Его легче всего получить декарбоксилированием пирослизевой кислоты (2,5-дикарбоксифурана), продукта пиролиза слизевой (тетрагидроксиадипиновой) кислоты. Наиболее общим методом получения фурановых производных является дегидратация g-дикетонов над хлоридом цинка:
Его легче всего получить декарбоксилированием пирослизевой кислоты (2,5-дикарбоксифурана), продукта пиролиза слизевой (тетрагидроксиадипиновой) кислоты. Наиболее общим методом получения фурановых производных является дегидратация g-дикетонов над хлоридом цинка:
 Сухая перегонка пентоз HOCH2(CHOH)3CHO дает фурфурол (a-формилфуран). Фурфурол проявляет многие свойства ароматического альдегида. Так, подобно бензальдегиду, он вступает в реакцию Канниццаро и в бензоиновую конденсацию.
Кумарон
(бензофуран) (см. выше «Конденсированные гетероциклические системы») вместе с его гомологами содержится в каменноугольной смоле. Он имеет некоторую ценность для получения кумароновых смол, которые образуются при его обработке серной кислотой. Производные кумарона могут быть получены разложением дибромкумаринов щелочью:
Сухая перегонка пентоз HOCH2(CHOH)3CHO дает фурфурол (a-формилфуран). Фурфурол проявляет многие свойства ароматического альдегида. Так, подобно бензальдегиду, он вступает в реакцию Канниццаро и в бензоиновую конденсацию.
Кумарон
(бензофуран) (см. выше «Конденсированные гетероциклические системы») вместе с его гомологами содержится в каменноугольной смоле. Он имеет некоторую ценность для получения кумароновых смол, которые образуются при его обработке серной кислотой. Производные кумарона могут быть получены разложением дибромкумаринов щелочью:
 или действием щелочи на о-гидрокси-b-хлорстирол, о-HO–C6H4–CH=CH–Cl. Кумароновая структура встречается во многих природных растительных веществах, которые являются мощными инсектицидами и ядами для рыб, например:
или действием щелочи на о-гидрокси-b-хлорстирол, о-HO–C6H4–CH=CH–Cl. Кумароновая структура встречается во многих природных растительных веществах, которые являются мощными инсектицидами и ядами для рыб, например:
 Тиофен
(формулу см. выше, т. кип. 84° С) содержится в каменноугольной смоле и сопровождает бензол при ее фракционировании. Его можно удалить из бензола осаждением комплекса с ацетатом ртути, из которого при обработке соляной кислотой можно регенерировать тиофен. Серная кислота также удаляет его из бензола путем образования a-тиофенсульфокислоты. Производные тиофена можно получить следующими способами:
Тиофен
(формулу см. выше, т. кип. 84° С) содержится в каменноугольной смоле и сопровождает бензол при ее фракционировании. Его можно удалить из бензола осаждением комплекса с ацетатом ртути, из которого при обработке соляной кислотой можно регенерировать тиофен. Серная кислота также удаляет его из бензола путем образования a-тиофенсульфокислоты. Производные тиофена можно получить следующими способами:
1) перегонкой янтарных кислот или g-кетокислот с P2S3:
 2) перегонкой g-дикетонов с P2S5:
2) перегонкой g-дикетонов с P2S5:
 Тиофен и его гомологи очень устойчивы к окислению или восстановлению кольца. Реакции ароматического замещения (сульфирование, нитрование и т.д.) идут в a-положение.
Тионафтен (бензотиофен) получают окислением о-меркаптокоричной кислоты красной кровяной солью (феррицианидом калия). Его 3-гидроксипроизводное, имеющее большое промышленное значение в химии красителей, получают действием уксусного ангидрида на о-карбоксифенилтиогликолевую кислоту о-HOOCC6H4–S–CH2COOH. Оно легко сочетается с солями диазония в положение 2, давая азокрасители, и конденсируется с альдегидами и кетонами, образуя тиоиндигоидные красители.
Тиофен и его гомологи очень устойчивы к окислению или восстановлению кольца. Реакции ароматического замещения (сульфирование, нитрование и т.д.) идут в a-положение.
Тионафтен (бензотиофен) получают окислением о-меркаптокоричной кислоты красной кровяной солью (феррицианидом калия). Его 3-гидроксипроизводное, имеющее большое промышленное значение в химии красителей, получают действием уксусного ангидрида на о-карбоксифенилтиогликолевую кислоту о-HOOCC6H4–S–CH2COOH. Оно легко сочетается с солями диазония в положение 2, давая азокрасители, и конденсируется с альдегидами и кетонами, образуя тиоиндигоидные красители.
 Пиррол (формулу см. выше), бесцветная, приятно пахнущая жидкость, содержащаяся в каменноугольной смоле, легко полимеризуется на воздухе. У него практически нет свойств основания, он устойчив к окислителям и щелочам, но легко полимеризуется в форме компонентов белков (пролин, триптофан), алкалоидов (никотин, атропин) и порфиринов (гемин, хлорофилл). Производные пиррола можно получить:
Пиррол (формулу см. выше), бесцветная, приятно пахнущая жидкость, содержащаяся в каменноугольной смоле, легко полимеризуется на воздухе. У него практически нет свойств основания, он устойчив к окислителям и щелочам, но легко полимеризуется в форме компонентов белков (пролин, триптофан), алкалоидов (никотин, атропин) и порфиринов (гемин, хлорофилл). Производные пиррола можно получить:
1) перегонкой сукцинимидов
 с цинковой пылью;
2) нагреванием g-дикетонов с аммиаком;
с цинковой пылью;
2) нагреванием g-дикетонов с аммиаком;
3) нагреванием слизевой кислоты ( см. выше) с аммиаком или первичными аминами;
4) одновременным восстановлением эквивалентных количеств b-кетоэфира и изонитрозокетона
 Пирролы вступают в типичные реакции ароматического замещения в a-положение. Обработка реактивов Гриньяра превращает их в a-пиррилмагнийгалогениды
Пирролы вступают в типичные реакции ароматического замещения в a-положение. Обработка реактивов Гриньяра превращает их в a-пиррилмагнийгалогениды
 которые вступают в типичные реакции Гриньяра.
Расширения кольца с образованием пиридиновой системы можно достичь:
которые вступают в типичные реакции Гриньяра.
Расширения кольца с образованием пиридиновой системы можно достичь:
1) обработкой хлороформом и этилатом натрия
 2) пропусканием a-алкилпирролов через трубку, нагретую до красного каления
2) пропусканием a-алкилпирролов через трубку, нагретую до красного каления
 Восстановление путем каталитического гидрирования под давлением, хотя и медленно, ведет к пирролидинам:
Восстановление путем каталитического гидрирования под давлением, хотя и медленно, ведет к пирролидинам:
 Индол
(бензопиррол; формулу см. в табл. 4, разд. III) содержится в каменноугольной смоле и эфирных маслах цветов апельсина и жасмина. Производные индола получают:
Индол
(бензопиррол; формулу см. в табл. 4, разд. III) содержится в каменноугольной смоле и эфирных маслах цветов апельсина и жасмина. Производные индола получают:
1) из о-аминофенилацетальдегида о-H2NC6H4CH2CH=O отщеплением воды;
2) нагреванием гидрохлоридов о-оў-диаминостильбенов:
 3) из фенилгидразонов нагреванием с галогенидами меди или цинка
3) из фенилгидразонов нагреванием с галогенидами меди или цинка
 По своим реакциям индол похож на пиррол с тем исключением, что в реакциях замещения участвует b-положение. Заслуживают упоминания следующие производные индола: 1) скатол (b-метилиндол), вещество с неприятным запахом, присутствующее в экскрементах; 2) триптофан (b-(b-индолил)аланин), аминокислота, встречающаяся во многих белках; 3) гетероауксин (b-индолилуксусная кислота или 3-индолилуксусная кислота), фактор роста растений; 4) индиго
По своим реакциям индол похож на пиррол с тем исключением, что в реакциях замещения участвует b-положение. Заслуживают упоминания следующие производные индола: 1) скатол (b-метилиндол), вещество с неприятным запахом, присутствующее в экскрементах; 2) триптофан (b-(b-индолил)аланин), аминокислота, встречающаяся во многих белках; 3) гетероауксин (b-индолилуксусная кислота или 3-индолилуксусная кислота), фактор роста растений; 4) индиго
 Оксазол
(формулу см. выше) известен в чистом виде. Его производные можно получить конденсацией амидов с a-галогенокетонами:
Оксазол
(формулу см. выше) известен в чистом виде. Его производные можно получить конденсацией амидов с a-галогенокетонами:
 или действием пентахлорида фосфора на ациламинокетоны:
или действием пентахлорида фосфора на ациламинокетоны:
 Оксазолы – слабые основания, чувствительные к расщеплению сильными кислотами.
Изоксазол
Оксазолы – слабые основания, чувствительные к расщеплению сильными кислотами.
Изоксазол
 и его производные представляют меньший интерес. Они могут быть получены дегидратацией монооксимов b-дикетонов.
Тиазол
и его гомологи – слабые основания, в которых кольцо обнаруживает высокую устойчивость к окислению, восстановлению и действию сильных кислот
и его производные представляют меньший интерес. Они могут быть получены дегидратацией монооксимов b-дикетонов.
Тиазол
и его гомологи – слабые основания, в которых кольцо обнаруживает высокую устойчивость к окислению, восстановлению и действию сильных кислот
 Тиазолы можно получить из a-ациламинокетонов действием P2S5, а также реакцией тиоамидов с a-галогенокетонами:
Тиазолы можно получить из a-ациламинокетонов действием P2S5, а также реакцией тиоамидов с a-галогенокетонами:
 Сильные кислоты превращают тиазолы в соли (C3H3SN + HX ® C3H3SNH+X–), которые устойчивы, но заметно гидролизуются в водных растворах. С алкилгалогенидами образуются N-замещенные соли тиазолия, содержащие четвертичный азот:
Сильные кислоты превращают тиазолы в соли (C3H3SN + HX ® C3H3SNH+X–), которые устойчивы, но заметно гидролизуются в водных растворах. С алкилгалогенидами образуются N-замещенные соли тиазолия, содержащие четвертичный азот:
 Наиболее важным природным соединением, содержащим тиазольное кольцо, является витамин B1 (тиамин). Ценный химиотерапевтический препарат сульфатиазол получают действием N-ацетилсульфанилхлорида на 2-аминотиазол с последующим удалением ацетильной группы гидролизом:
Наиболее важным природным соединением, содержащим тиазольное кольцо, является витамин B1 (тиамин). Ценный химиотерапевтический препарат сульфатиазол получают действием N-ацетилсульфанилхлорида на 2-аминотиазол с последующим удалением ацетильной группы гидролизом:
 Имидазол
(глиоксалин) и его гомологи
Имидазол
(глиоксалин) и его гомологи
 получают из альдегидов, a-дикетонов и аммиака:
получают из альдегидов, a-дикетонов и аммиака:
 Их также можно приготовить взаимодействием амидинов
Их также можно приготовить взаимодействием амидинов
 с a-галогенокетонами. Имидазолы – более сильные основания, чем пирролы. С алкилгалогенидами они дают N-алкилимидазолы. Эти вещества при пропускании через трубку при температуре красного каления изомеризуются в 2-алкилимидазолы; при взаимодействии со второй молекулой алкилгалогенида они превращаются в соли имидазолия, содержащие четвертичный азот
с a-галогенокетонами. Имидазолы – более сильные основания, чем пирролы. С алкилгалогенидами они дают N-алкилимидазолы. Эти вещества при пропускании через трубку при температуре красного каления изомеризуются в 2-алкилимидазолы; при взаимодействии со второй молекулой алкилгалогенида они превращаются в соли имидазолия, содержащие четвертичный азот
 Действие реактивов Гриньяра RMgX на имидазолы ведет к соответствующим 2-имидазолилмагнийгалогенидам C3H3N2MgX, которые вступают в реакции, обычные для реактивов Гриньяра. Имидазольное кольцо встречается во многих природных соединениях, в том числе в аминокислоте гистидине (см. разд. IV-1.Б.4, «Аминокислоты»), алкалоидах группы пилокарпина и пуриновых основаниях.
Пиразол
и его производные – только синтетические соединения; кольцевая система пиразола
Действие реактивов Гриньяра RMgX на имидазолы ведет к соответствующим 2-имидазолилмагнийгалогенидам C3H3N2MgX, которые вступают в реакции, обычные для реактивов Гриньяра. Имидазольное кольцо встречается во многих природных соединениях, в том числе в аминокислоте гистидине (см. разд. IV-1.Б.4, «Аминокислоты»), алкалоидах группы пилокарпина и пуриновых основаниях.
Пиразол
и его производные – только синтетические соединения; кольцевая система пиразола
 не встречается в природе. Пиразолы получают взаимодействием гидразина с b-дикетонами:
не встречается в природе. Пиразолы получают взаимодействием гидразина с b-дикетонами:
 или действием диазоалканов на ацетилен:
или действием диазоалканов на ацетилен:
 Реакция фенилгидразина с a,b-ненасыщенными кетонами или эфирами дает дигидропиразолы, или пиразолины:
Реакция фенилгидразина с a,b-ненасыщенными кетонами или эфирами дает дигидропиразолы, или пиразолины:
 Эти соединения легко окисляются в соответствующие пиразолы. Пиразольное кольцо очень устойчиво к окислению, восстановлению и действию сильных кислот. Пиразолиниевые соли, получаемые действием сильных кислот на пиразолины, нестойки и разлагаются в вакууме.
Наиболее важный класс пиразолов – пиразолоны
Эти соединения легко окисляются в соответствующие пиразолы. Пиразольное кольцо очень устойчиво к окислению, восстановлению и действию сильных кислот. Пиразолиниевые соли, получаемые действием сильных кислот на пиразолины, нестойки и разлагаются в вакууме.
Наиболее важный класс пиразолов – пиразолоны
 получаемые действием гидразина и его производных на b-кетоэфиры, например,
получаемые действием гидразина и его производных на b-кетоэфиры, например,
 Пиразолоны ведут себя как смесь трех таутомерных (т.е. находящихся в равновесии) форм, например:
Пиразолоны ведут себя как смесь трех таутомерных (т.е. находящихся в равновесии) форм, например:
 1-Фенил-3-метилпиразол-5 является важным веществом. Окисление красной кровяной солью (феррицианидом калия) превращает его в индигоидный краситель пиразоловый голубой:
1-Фенил-3-метилпиразол-5 является важным веществом. Окисление красной кровяной солью (феррицианидом калия) превращает его в индигоидный краситель пиразоловый голубой:
 Метилирование (CH3I при 100° С) превращает его в жаропонижающий препарат антипирин (1-фенил-2,3-диметилпиразолон), 4-N-диметиламинопроизводное которого представляет собой аналогичное лекарственное средство амидопирин (пирамидон).
Кольцевые системы с тремя и более гетероатомами не представляют практического интереса. Все они устойчивы к окислению, восстановлению и действию сильных кислот. Фуразаны получают дегидратацией диоксимов a-дикетонов. 1,2,3-Триазолы и тетразолы также относятся к этой группе соединений.
ЛИТЕРАТУРА
Фризер Л., Фризер М. Органическая химия. Углубленный курс. М., 1972
Метилирование (CH3I при 100° С) превращает его в жаропонижающий препарат антипирин (1-фенил-2,3-диметилпиразолон), 4-N-диметиламинопроизводное которого представляет собой аналогичное лекарственное средство амидопирин (пирамидон).
Кольцевые системы с тремя и более гетероатомами не представляют практического интереса. Все они устойчивы к окислению, восстановлению и действию сильных кислот. Фуразаны получают дегидратацией диоксимов a-дикетонов. 1,2,3-Триазолы и тетразолы также относятся к этой группе соединений.
ЛИТЕРАТУРА
Фризер Л., Фризер М. Органическая химия. Углубленный курс. М., 1972
Общая органическая химия. М., 1981–1985
Марч Д. Органическая химия. Реакции, механизмы и структура, тт. 1–4. М., 1987–1988
|